Table of Contents
- Introduction
- Folate and Carcinogenesis: A Dual-Edged Metabolic Sword
- Mechanistic Pathways and Modifying Factors in Folate-Driven Tumor Biology
- Folate’s Protective Role in DNA Metabolism and Cancer Prevention
- Uracil-Induced DNA Damage and Modifying Nutritional Factors
- Genomic Vulnerability: Site-Specific DNA Damage Under Folate Stress
- Uracil Misincorporation: A Driver of Mutation and Structural Instability
- Human Trials and Biomarkers of Chromosomal Damage
- Next Steps: Methylation and Beyond
- Take-Home Messages
- Summary and Conclusions
- Did You Know About Folate Receptor Autoantibodies (FRAAs) and Brain Development?
- References
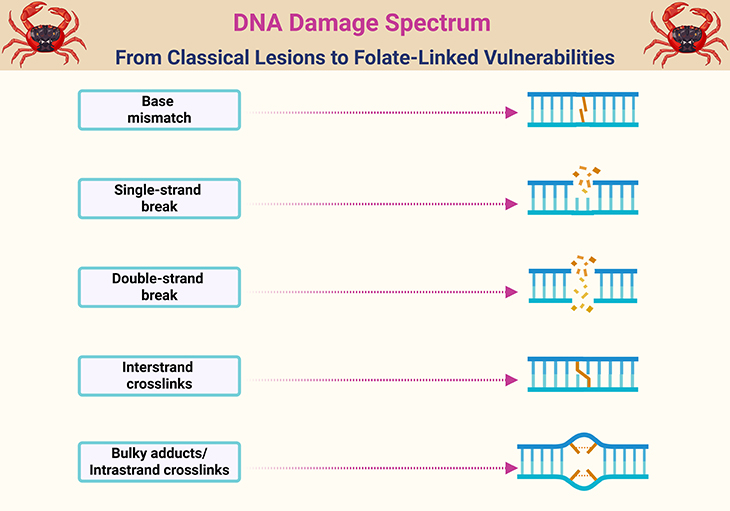
Figure 1. DNA Damage Spectrum: From Classical Lesions to Folate-Linked Vulnerabilities. Illustration of major DNA lesions affecting genomic integrity, including (1) base mismatches arising from replication errors, (2) single-stranded breaks caused by oxidative stress or repair intermediates, (3) double-stranded breaks resulting from replication fork collapse or enzymatic cleavage, (4) interstrand cross-links that obstruct strand separation during replication, and (5) bulky adducts or intrastrand crosslinks induced by environmental mutagens or metabolic byproducts. Folate deficiency or impaired one-carbon metabolism can contribute to each of these damage types through distinct biochemical pathways: (A) Base mismatch: Elevated UTP/TTP ratio leads to uracil misincorporation in place of thymidine, resulting in frequent base mismatches and repair stress. (B) Single-stranded breaks: Excision of uracil by base excision repair enzymes creates abasic sites and transient single-strand breaks, especially in rapidly dividing cells. (C) Double-stranded breaks: Closely spaced uracil lesions on opposite strands or failed repair of single-strand breaks can culminate in double-strand breaks, particularly within tumor suppressor loci (Apc, p53). (D) Interstrand cross-links: Folate-related methylation defects may impair repair pathways (e.g., Fanconi anemia pathway), increasing susceptibility to cross-linking agents. (E) Bulky adducts/intrastrand crosslinks: Aberrant methylation patterns and impaired detoxification may enhance vulnerability to endogenous or exogenous adduct formation, compounding genomic instability. These aberrations underscore folate’s role as a genomic gatekeeper—where deficiency, excess, or dysregulation can tip the balance from repair to rupture, with implications for both cancer risk and therapeutic safety.
Introduction
Folate, a cornerstone of one-carbon metabolism, has long been celebrated for its indispensable role in DNA synthesis, repair, and methylation—processes that safeguard genomic integrity and regulate gene expression. As a dietary micronutrient and therapeutic agent, folate occupies a unique position at the intersection of nutritional biochemistry, developmental biology, and cancer prevention. Yet, as our understanding deepens, so too does the complexity of its narrative.
Over the past two decades, a robust body of epidemiological, clinical, and mechanistic evidence has converged to support the notion that adequate folate status reduces the risk of several cancers, most notably those of the colorectum, breast, and pancreas. The biological rationale is compelling: folate deficiency disrupts nucleotide balance, promotes uracil misincorporation, impairs DNA repair, and alters methylation patterns—each a potential trigger for malignant transformation. However, this protective paradigm is now tempered by a growing recognition of folate’s context-dependent duality.
In individuals with pre-existing neoplastic lesions, high-dose folate—particularly in the form of folic acid or folinic acid—may paradoxically accelerate tumor progression by fueling the proliferative demands of dysplastic cells. This phenomenon, once viewed as paradoxical, is now understood as a predictable consequence of folate’s biochemical potency. It is within this nuanced framework that public health recommendations, clinical interventions, and precision nutrition strategies must be re-evaluated.
Nowhere is this tension more palpable than in the autism community, where high-dose folinic acid is increasingly used to support neurodevelopmental outcomes in children with cerebral folate deficiency or mitochondrial dysfunction. While many families report meaningful improvements, a lingering concern persists: could prolonged exposure to supraphysiologic folate levels—especially in genetically or metabolically vulnerable children—carry unintended oncogenic risks? Although definitive answers remain elusive, the question underscores the urgent need for longitudinal safety data, biomarker surveillance, and a mechanistic understanding of how folate modulates cellular fate across developmental and disease contexts.
This article synthesizes the current landscape of folate and carcinogenesis, with a focus on the molecular mechanisms, genomic vulnerabilities, and nutrient-gene interactions that shape cancer risk. In doing so, it aims to inform a more nuanced, evidence-based dialogue—one that honors both the promise and the complexity of folate in human health.
Folate and Carcinogenesis: A Dual-Edged Metabolic Sword
A substantial and steadily expanding body of preclinical and clinical evidence, particularly in the context of colorectal neoplasia, supports a protective role for folate in cancer prevention. This protective association is also emerging—albeit with less consistency—for malignancies of the breast, lung, pancreas, and esophagus. However, this narrative is complicated by a paradoxical observation: when folate is consumed in excessive amounts, especially in individuals who already harbor precancerous or malignant lesions, it may accelerate tumor progression rather than prevent it.
What initially appears to be a contradiction is, in fact, biologically coherent when viewed through the lens of folate’s cellular functions. This “dual effect” hypothesis—where folate may either suppress or promote carcinogenesis depending on timing and cellular context—poses a significant challenge for public health policy, as it implies that population-wide recommendations may not be universally beneficial. Thus, any mechanistic framework aiming to explain folate’s role in cancer risk must grapple with this bidirectional potential.
Mechanistic Pathways and Modifying Factors in Folate-Driven Tumor Biology
At the molecular level, folate’s sole established biochemical role is its function as a cofactor and substrate in one-carbon (1C) metabolism—a network of reactions that transfers single-carbon units between molecules. Through this role, folate is indispensable for both biological methylation and the de novo synthesis of purines and thymidylate, which are essential for DNA replication and repair.
The integrity of DNA synthesis, repair, and methylation—all of which are tightly folate-dependent—is critical for maintaining genomic stability. Disruptions in these processes are well-recognized hallmarks of malignant transformation. Consequently, folate deficiency is theorized to promote carcinogenesis by fostering uracil misincorporation, DNA strand breaks, and global hypomethylation, all of which can lead to oncogene activation or tumor suppressor gene silencing (see Figure 1) [1-2].
Conversely, in rapidly proliferating neoplastic cells, which have elevated demands for nucleotide synthesis, excess folate may inadvertently fuel tumor growth—particularly when administered after neoplastic transformation has already begun. This paradox underscores the importance of timing, dosage, and cellular context in determining folate’s net effect on cancer risk.
Adding further complexity, several modifying factors influence folate’s impact on carcinogenesis. These include:
- Habitual alcohol consumption, which impairs folate absorption and metabolism
- Age, which may alter methylation dynamics and DNA repair capacity
- Genetic polymorphisms (e.g., in MTHFR) that affect folate utilization
- Micronutrient status within the broader one-carbon network, including:
- Vitamin B2 (riboflavin) – cofactor for MTHFR
- Vitamin B6 (pyridoxal phosphate) – involved in transsulfuration
- Vitamin B12 (cobalamin) – essential for methionine synthase
- Methionine and choline – methyl donors and intermediates
The biochemical interdependence of these nutrients is well-established, and their collective influence on methylation and nucleotide synthesis is increasingly recognized as clinically significant. Notably, population-based surveys reveal that subclinical deficiencies—nutrient levels below optimal but not low enough to cause overt deficiency syndromes—are surprisingly prevalent in adults across industrialized nations. This is particularly true for vitamins B₆ and B₁₂, whose inadequacies have been linked in multiple studies to elevated cancer risk.
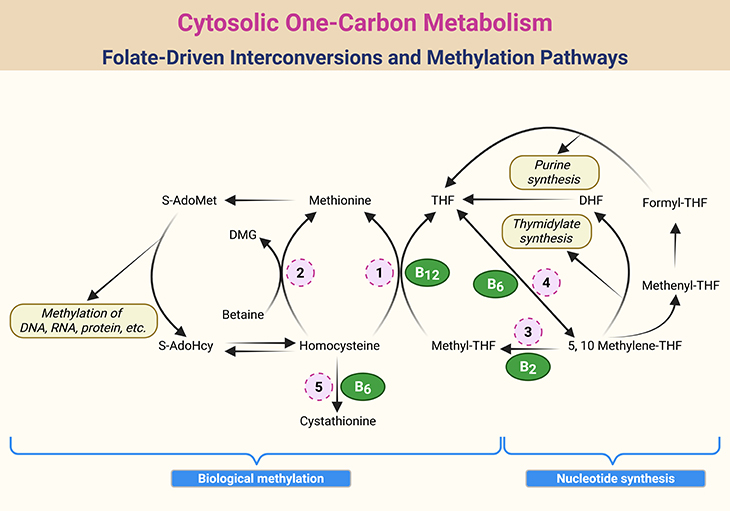
Figure 2. Cytosolic One-Carbon Metabolism: Folate-Driven Interconversions and Methylation Pathways. Schematic representation of one-carbon metabolism within the cytosol of mammalian cells, highlighting the dynamic interconversion of folate coenzymes and their roles in nucleotide synthesis and methylation. Key intermediates include tetrahydrofolate (THF), dihydrofolate (DHF), S-adenosylmethionine (S-AdoMet), S-adenosylhomocysteine (S-AdoHcy), and dimethylglycine (DMG). Enzymatic steps are catalyzed by: 1. Methionine synthase (MS); 2. Betaine-homocysteine methyltransferase (BHMT); 3. Methylenetetrahydrofolate reductase (MTHFR); 4. Serine hydroxymethyltransferase (SHMT); 5. Cystathionine β-synthase (CBS).
Folate’s Protective Role in DNA Metabolism and Cancer Prevention
The foundational role of folate in DNA metabolism—both at the genetic and epigenetic levels—serves as the logical entry point for understanding its potential to protect against cancer (see Figure 2). Aberrations in DNA synthesis, repair, and methylation are widely recognized as critical pathways in carcinogenesis, and folate is intimately involved in all three. Given the diverse enzymatic functions of folate derivatives, it is plausible that its protective effects arise from a convergence of multiple biochemical pathways, rather than a single mechanism.
One of the most studied mechanisms involves uracil misincorporation and its impact on genomic integrity (see Figure 3). Folate, specifically in the form of 5,10-methylenetetrahydrofolate (5,10-methylene THF), is essential for the conversion of uracil to thymidine—a key step in DNA synthesis. When folate availability is limited, the intracellular UTP/TTP ratio increases, leading to the erroneous incorporation of uracil into DNA. This phenomenon has been consistently demonstrated in cell culture, animal models, and human studies under conditions of severe folate deficiency. However, it remains uncertain whether individuals with marginal folate status—common in the general population—experience sufficient disruption in de novo thymidine synthesis to cause significant uracil accumulation in DNA.
Uracil-Induced DNA Damage and Modifying Nutritional Factors
The susceptibility of tissues to uracil misincorporation is not governed by folate status alone. In rodent models, advanced age was shown to increase the colon’s vulnerability to uracil incorporation under folate-deficient conditions. Furthermore, vitamin B₁₂ deficiency—which induces a functional folate deficiency via the methyl-folate trap—has been linked to elevated uracil levels in colonic DNA, even in the absence of overt folate depletion. In a human intervention study, individuals with low baseline plasma B₁₂ (<400 pg/mL) exhibited a blunted reduction in leukocyte uracil following folic acid supplementation, underscoring the interdependence of B₁₂ and folate in maintaining DNA integrity [3-4].
Interestingly, a mild depletion of all four one-carbon nutrients—B₂, B₆, B₁₂, and folate—did not significantly increase uracil misincorporation in the mouse colon, suggesting that threshold effects or nutrient synergy may modulate this response.
Under normal physiological conditions, low levels of uracil in DNA are tolerated and efficiently repaired. However, when uracil incorporation becomes excessive, it triggers a cascade of genetic instability. Mammalian cells deploy base excision repair (BER) systems to remove uracil residues, a process that first creates an abasic site, followed by a single-strand break in the DNA backbone. When these breaks occur on opposite strands within ~14 base pairs, they culminate in double-stranded breaks (DSBs)—a highly mutagenic event (see Figure 1 and Figure 3).
DSBs are implicated in:
- Tumor suppressor gene deletions
- Chromosomal translocations
- Oncogene amplification
- Transcriptional silencing due to impaired RNA polymerase progression
Animal studies have demonstrated that folate depletion induces strand breaks in the coding regions of Apc and p53 genes, accompanied by reduced mRNA expression of these critical tumor suppressors. While causality between strand breaks and transcriptional repression remains to be definitively proven, the correlation is compelling. Notably, in cultured human lymphocytes, p53 strand breaks did not consistently lead to reduced p53 mRNA, suggesting that gene-specific repair dynamics or compensatory transcriptional mechanisms may influence outcomes.
Genomic Vulnerability: Site-Specific DNA Damage Under Folate Stress
While folate deficiency—and broader depletion of one-carbon nutrients—has been shown to induce genome-wide DNA strand breaks, emerging evidence suggests that this damage is not uniformly distributed. Instead, certain genomic regions appear disproportionately vulnerable. Even within a single gene, strand break susceptibility varies by locus. In rodent models, the colonic Apc and p53 genes exhibited damage concentrated in highly conserved regions—the same regions most frequently mutated in human cancers.
Specifically:
- In the Apc gene, strand breaks clustered in the “mutation cluster region” of exon 15
- In p53, damage localized to exons 5–8, the so-called “hypermutable region” where ~90% of p53 mutations in human cancers occur
Moreover, concurrent depletion of folate, B₂, B₆, and B₁₂—even at subclinical levels—amplifies strand breakage beyond what is observed with folate deficiency alone. This synergistic effect underscores the interconnectedness of the one-carbon network (see Figure 2) and suggests that even mild inadequacies can have compounding genomic consequences.
One intuitive hypothesis is that site-specific uracil incorporation may define a region’s susceptibility to strand breaks. However, technological limitations currently prevent accurate mapping of uracil incorporation at the nucleotide level, leaving this theory untested.
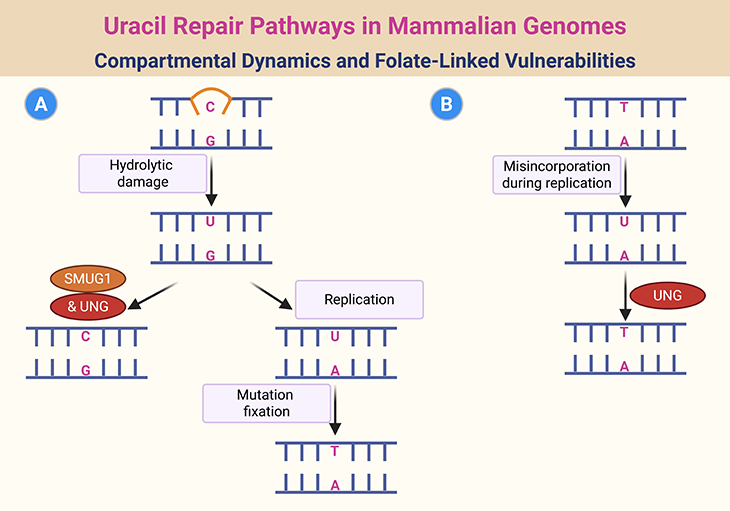
Figure 3. Uracil Repair Pathways in Mammalian Genomes: Compartmental Dynamics and Folate-Linked Vulnerabilities. Illustration of the compartmentalized mechanisms for repairing uracil residues in mammalian DNA. Regions of DNA transiently exposed in single-stranded form during replication or transcription are prone to hydrolytic deamination of cytosine, generating U:G mismatches (Pathway A). These lesions are recognized and excised by SMUG1 uracil-DNA glycosylase. If left unrepaired, they persist as U:A base pairs in subsequent replication cycles, leading to C→T transition mutations. Separately, UNG uracil-DNA glycosylase primarily targets U:A base pairs that arise from the misincorporation of dUTP in place of TTP during DNA synthesis—a process exacerbated under folate-deficient conditions (Pathway B). Additional uracil glycosylases encoded by TDG and MBD4 contribute to repair, though their specific roles remain less clearly defined.
Uracil Misincorporation: A Driver of Mutation and Structural Instability
Uracil incorporation into DNA can occur via two distinct mechanisms, each with unique consequences (see Figure 3):
- Spontaneous deamination of cytosine produces a U:G mispair, a nonenzymatic event estimated to occur 100–500 times per cell per day. If replication precedes repair, this leads to a C:G → T:A transition mutation, a common mutational signature in cancer.
- Folate deficiency, on the other hand, promotes uracil insertion where thymidine is normally required, resulting in a U:A mispair. While not directly mutagenic, this mispair may distort DNA’s secondary structure, disrupting the function of chromatin-associated proteins that rely on precise DNA conformation.
Experimental folate deficiency in rodents has also been shown to impair excision repair in colonic tissue, though whether this is due to excess uracil or other metabolic disruptions remains unclear.
Collectively, these findings have positioned excessive uracil incorporation as a central hypothesis in explaining how inadequate folate status promotes carcinogenesis. While aberrant DNA methylation remains a plausible contributor, recent data suggest that defects in DNA synthesis and repair may be even more critical.
In comparative studies of mouse strains:
- BALB/C mice, which are more susceptible to intestinal tumors, exhibited:
- Elevated UTP/TTP ratios
- Increased double-stranded DNA breaks
- Greater overall DNA damage
- In contrast, C57/B16 mice, which are more tumor-resistant, showed significantly less damage under similar folate-depleted conditions
Importantly, restoring methylation in BALB/C mice using trimethylglycine (betaine) did not reduce tumorigenesis, suggesting that nucleotide synthesis impairment, not methylation loss, may be the dominant driver.
Further evidence comes from studies using rodents with gain- or loss-of-function mutations in cytosolic serine hydroxymethyltransferase (cSHMT)—a key enzyme that governs the partitioning of folate coenzymes between methylation and nucleotide synthesis. Tumor susceptibility correlated with loss of coenzyme availability for nucleotide synthesis, but not with loss of methylation substrate. Intriguingly, folate depletion in colon cancer cells can alter cSHMT expression, introducing yet another mechanism by which folate imbalance may disrupt normal metabolic partitioning [5-6].
Human Trials and Biomarkers of Chromosomal Damage
Human intervention trials have begun to clarify the conditions under which one-carbon vitamin supplementation can mitigate uracil incorporation and chromosomal instability. The latter is often assessed using the micronucleus index, a biomarker reflecting chromosomal breaks and losses and considered a predictive marker of cancer risk.
Key findings include:
- Folic acid supplementation consistently reduces uracil content in peripheral blood DNA
- However, reductions in chromosomal damage metrics are only observed in individuals with elevated baseline damage, typically due to folate depletion or related factors
- In one trial, low plasma B₁₂ (<400 pg/mL) blunted the uracil-lowering effect of folic acid, reinforcing the importance of B₁₂ status
- In contrast, a 6-month intervention using 5 mg folic acid + 1.25 mg B₁₂ failed to reduce uracil levels in colonic mucosa (van den Donk et al. 2007) [6], suggesting tissue-specific responses or unique colonic dynamics
Next Steps: Methylation and Beyond
While uracil incorporation and DNA repair have emerged as dominant mechanisms linking folate to carcinogenesis, other pathways—particularly those involving biological methylation—remain critical and will be explored in depth in the next installment of this series.
Take-Home Messages
- Folate’s role in carcinogenesis is context-dependent—protective in early stages, potentially permissive in established neoplasia. Timing, dose, and cellular state matter.
- Uracil misincorporation is not merely a biochemical curiosity—it is a mechanistic gateway to genomic instability, strand breaks, and mutational cascades.
- DNA damage is not random—folate depletion targets conserved, mutation-prone regions of tumor suppressor genes like Apc and p53, amplifying oncogenic risk.
- The one-carbon network is a metabolic symphony—folate, B₂, B₆, B₁₂, methionine, and choline act in concert. Subclinical deficiencies in any can tip the balance toward instability.
- Micronutrient synergy matters—mild inadequacies across multiple one-carbon nutrients can produce effects more profound than folate depletion alone.
- Methylation is not the whole story—recent evidence suggests that impaired nucleotide synthesis and repair may be the more decisive drivers of tumorigenesis under folate stress.
- Genetic background shapes vulnerability—strain-specific differences in mice (e.g., BALB/C vs. C57/B16) reveal how folate status interacts with inherent repair capacity and tumor susceptibility.
- Human trials underscore complexity—folic acid supplementation reduces uracil in blood cells, but not always in colonic mucosa, hinting at tissue-specific dynamics and the need for precision nutrition.
Cf. previous blogs entitled as: “MTHFR at the Crossroads: Genetic Variants, Metabolic Disruption, and Clinical Consequences.”; “Cracking the Folate Code: How Enzymatic Polymorphisms Shape Health and Neurodevelopment.”)
Summary and Conclusions
The intricate relationship between folate status and carcinogenesis continues to unfold with both promise and complexity. Central to this narrative is the mechanism of uracil misincorporation into DNA, a consequence of impaired thymidylate synthesis under conditions of folate deficiency. This biochemical disruption elevates the intracellular UTP/TTP ratio, leading to the erroneous incorporation of uracil in place of thymidine—a process that triggers single- and double-stranded DNA breaks, particularly within mutation-prone regions of tumor suppressor genes such as Apc and p53. These strand breaks compromise genomic integrity, promote mutational events, and may suppress gene expression, thereby laying the groundwork for malignant transformation. The susceptibility to such damage is further amplified by subclinical deficiencies in other one-carbon nutrients—B₂, B₆, B₁₂, and choline—highlighting the synergistic nature of the one-carbon metabolic network.
Importantly, recent animal studies suggest that defects in nucleotide synthesis and repair may play a more decisive role in tumorigenesis than disruptions in methylation alone. This insight challenges long-held assumptions and calls for a recalibration of mechanistic models. Moreover, emerging data from human intervention trials reveal tissue-specific responses to folate supplementation, with colonic mucosa showing resistance to uracil reduction despite systemic improvements—underscoring the need for precision nutrition and biomarker-guided strategies.
Despite these advances, critical gaps remain. The inability to accurately map site-specific uracil incorporation limits our understanding of genomic vulnerability. The long-term consequences of high-dose folinic acid therapy, particularly in pediatric populations with neurodevelopmental disorders, remain insufficiently studied. As folate continues to be deployed therapeutically in diverse clinical contexts—from cancer prevention to autism management—there is an urgent need for longitudinal safety data, mechanistic studies, and population-specific risk assessments.
In conclusion, folate’s role in carcinogenesis is neither linear nor universal. It is a context-dependent modulator of cellular fate, capable of both protecting and promoting depending on timing, dosage, and molecular milieu. Future research must embrace this complexity, integrating genetic background, nutrient interactions, and tissue-specific dynamics to guide safe and effective interventions.
Did You Know? Folate Receptor Autoantibodies (FRAAs) may impede proper folate transport.
Folate (vitamin B9) is very important for your child’s brain development!
During pregnancy, it helps prevent neural tube defects and plays a big role in forming a normal and healthy baby’s brain and spinal cord. Folate also helps cells divide and assists in both DNA and RNA synthesis.
Emerging research suggests that the presence of FRAAs negatively impacts folate transport into the brain.
- Recent studies reveal that a large subgroup of children with autism spectrum disorder (ASD) have FRAAs.
- This suggests that a possible disruption in folate transport across the blood-cerebrospinal fluid (CSF) barrier may potentially influence ASD-linked brain development.
- Screening for the FRAAs in your child should be part of your early intervention strategies.
Is there a test for identifying Folate Receptor Autoantibodies (FRAAs)?
Yes, there is a test – The Folate Receptor Antibody Test (FRAT®) has emerged as a diagnostic tool for detecting the presence of FRAAs.
It is important to screen at an early age or as soon as possible as there may be corrective measures available. Please consult your physician for further information.
To request a test kit, click on the button below.

For information on autism monitoring, screening and testing please read our blog.
References
- Blount BC, Mack MM, Wehr CM, MacGregor JT, Hiatt RA, Wang G, Wickramasinghe SN, Everson RB, Ames BN. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci U S A. 1997 Apr 1;94(7):3290-5. doi: 10.1073/pnas.94.7.3290. PMID: 9096386; PMCID: PMC20362.
https://pubmed.ncbi.nlm.nih.gov/9096386/
→ Demonstrates how folate deficiency leads to massive uracil incorporation and chromosomal breaks in human cells. - Duthie SJ. Folate and cancer: how DNA damage, repair and methylation impact on colon carcinogenesis. J Inherit Metab Dis. 2011 Feb;34(1):101-9. doi: 10.1007/s10545-010-9128-0. Epub 2010 Jun 11. PMID: 20544289.
https://pubmed.ncbi.nlm.nih.gov/20544289/
→ Reviews folate’s dual role in DNA stability and its context-dependent influence on colon cancer risk. - Duthie SJ, Grant G, Narayanan S. Increased uracil misincorporation in lymphocytes from folate-deficient rats. Br J Cancer. 2000 Dec;83(11):1532-7. doi: 10.1054/bjoc.2000.1481. PMID: 11076664; PMCID: PMC2363426.
https://pubmed.ncbi.nlm.nih.gov/11076664/
→ Animal study showing increased uracil misincorporation and DNA strand breaks under moderate folate deficiency. - Kim YI. Role of folate in colon cancer development and progression. J Nutr. 2003 Nov;133(11 Suppl 1):3731S-3739S. doi: 10.1093/jn/133.11.3731S. PMID: 14608107.
https://pubmed.ncbi.nlm.nih.gov/14608107/
→ Explores folate’s protective and potentially tumor-promoting effects depending on timing and dose. - MacFarlane AJ, Liu X, Perry CA, Flodby P, Allen RH, Stabler SP, Stover PJ. Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J Biol Chem. 2008 Sep 19;283(38):25846-53. doi: 10.1074/jbc.M802671200. Epub 2008 Jul 21. PMID: 18644786; PMCID: PMC2533799.
https://pubmed.ncbi.nlm.nih.gov/18644786/
→ Highlights that mitochondrial SHMT-derived one-carbon units are essential for folate-mediated one-carbon metabolism in the cytoplasm. - van den Donk M, Pellis L, Crott JW, van Engeland M, Friederich P, Nagengast FM, van Bergeijk JD, de Boer SY, Mason JB, Kok FJ, Keijer J, Kampman E. Folic acid and vitamin B-12 supplementation does not favorably influence uracil incorporation and promoter methylation in rectal mucosa DNA of subjects with previous colorectal adenomas. J Nutr. 2007 Sep;137(9):2114-20. doi: 10.1093/jn/137.9.2114. PMID: 17709451.
https://pubmed.ncbi.nlm.nih.gov/17709451/
→ Human trial showing tissue-specific response to folate/B12 supplementation, with no reduction in colonic uracil levels.


