Table of Contents
- Introduction
- Epigenetic Layers and Players
- 1. DNA Methylation
- 2. Histone Variants and Histone Modifications
- 3. Non-coding RNAs
- Time Points of Plasticity in the Epigenome
- Take Home Messages
- Summary and Conclusions
- Did You Know About Folate Receptor Autoantibodies (FRAAs) and Brain Development?
- References
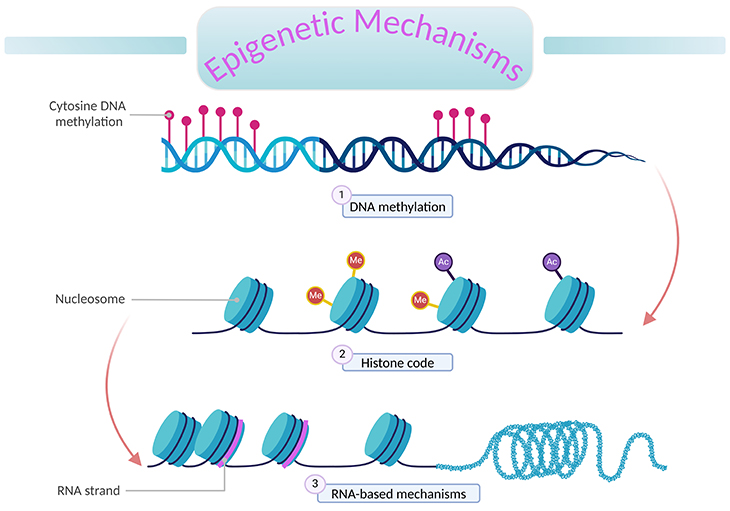
Figure 1. An overview of epigenetic mechanisms. ‘Epigenetics’ literally means ‘above DNA.’ A genome is the complete set of genetic information (DNA) in an organism. Epigenetic processes describe changes to the genome that can alter gene expression without changing the underlying DNA sequence. These changes are mitotically inheritable; and involve the interplay of (1) DNA methylation, (2) histone (H2A, H2B, H3, and H4) modification, and (3) RNA-based mechanisms (non-coding RNA molecules).
Introduction
The field of epigenetics has evolved rapidly over recent decades; and generated a great deal of interest, primarily focusing on how our very molecular makeup in the form of modification to the genome can be altered by factors as varied as nutrition, aging, disease, stress, alcohol, and exposure to pollutants [1].
Epigenetic changes have previously been implicated in the etiology of a variety of diseases, for example, in the initiation and progression of certain cancers, and inherited growth disorder syndromes. However, the exploration of epigenetics’ role in fetal reprograming is still in its infancy.
This series of blogs focuses on how nutritional exposure during pregnancy may influence the infant epigenome, and the meaningful impact that such modifications may have on the long-term health of the child. In order to better comprehend:
- First, in this blog, we start by describing some key concepts in epigenetics and discuss windows of epigenetic plasticity in the context of the ‘developmental origins of health and disease (DOHaD)’ hypothesis – the concept that early life ‘exposure’ influence lifelong health.
- Second, in subsequent blogs, we present some of the key mechanisms by which nutrition can affect the epigenome, with a particular focus on the role of one-carbon (1-C) metabolism, specifically folate and DNA methylation.
- Next, we finish by outlining some of the child health outcomes that have been linked to epigenetic dysregulation, mainly focusing on epigenome, neurodevelopment, and autism spectrum disorders (ASDs).
- Finally, we touch upon possible next steps that need to be realized if insights into the basic science of epigenetics are to be translated into tangible public health benefits.
Epigenetic Layers and Players
Epigenetic processes describe changes to the genome that can alter gene expression without changing the underlying DNA sequence. These changes are mitotically heritable, and involve the interplay of the following chromatin and epigenetic mechanisms (see Figure 1), for example:
- DNA methylation,
- histone modification and histone variants,
- chromatin loops,
- chromatic modifying proteins, and
- non-coding RNA molecules.
1. DNA Methylation
DNA methylation most commonly occurs at loci where a cytosine is found next to a guanine on a DNA strand along its linear sequence, consequently termed cytosine-phosphate-guanine or CpG sites. It involves the covalent bonding of a methyl (CH3) group to the cytosine at the 5’-carbon position to form 5-methylcytosine [2].
CpGs found in high densities are termed CpG islands. Approximately two-thirds of human genes contain CpG islands in their promoter regions, although repetitive elements in the genome can also contain many CpG sites. CpGs are generally methylated in non-promoter regions and unmethylated at promoter regions. Most importantly, methylation at CpG sites in promoters is usually associated with transcriptional silencing, although not consistently.
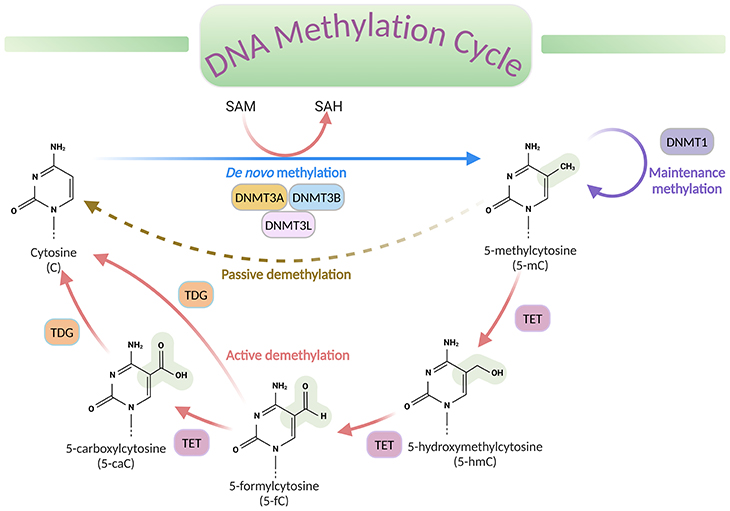
Figure 2. DNA methylation cycle. DNA methylation involves the transfer of a methyl group (CH3) from the universal donor S-adenosylmethionine (SAM) to the fifth carbon of a cytosine (C), by a DNA methyltransferase (DNMT), to produce 5-methylcytosine (5-mC). S-adenosylhomocysteine (SAH) is produced as a byproduct. DNA de-methylation occurs via passive or active processes that may co-exist in cells. Active de-methylation involves the sequential conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC) and derivatives of 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC), likely then modified to cytosine (C); and it is regulated by ten-eleven translocation (TET) family of proteins.[DNMT, DNA methyltransferase; TET, ten-eleven translocation proteins; TDG, thymine DNA glycosylase]
The process of DNA methylation universally involves the transfer of a methyl (CH3) group from S-adenosyl-L-methionine (SAM or AdoMet) to cytosine, catalyzed by DNA methyltransferase enzymes (DNMTs), to produce 5-methylcytosine (5-mC) (see Figure 2).
Even though methylation is the most examined chemical alteration till date, other less abundant forms of modifications, such as 5-hydroxymethlcytosine (5-hmC), 5-formylcytosine (5-fC), and 5-carboxycytosine (5-caC) have now been described in specific contexts. These variants are now considered to be intermediates within a ‘DNA methylation cycle’ that involves both the addition and the removal of methylation from DNA (see Figure 2).
N6-methyladenosine (m6A) or N6-methyl-2’-deoxyadenosine (m6dA) has also been described as a modification of mRNA, tRNA, and other non-coding RNAs, this is in addition to a subset of tissue-/developmental-specific genomic DNA. The mRNA m6A modification has been widely characterized and plays a critical role in several developmental processes, while limited information on DNA m6A is currently available.
DNA methylation is catalyzed by DNA methyltransferase (DNMTs). In mammals, the family of cytosine DNMTs includes DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L. Whereas the following three DNMTs (viz., DNMT1, DNMT3A, and DNMT3B) are canonical cytosine-5-methyltransferases that catalyze the addition of methylation marks to DNA, DNMT2 and DNMT3L do not. The corresponding writers of m6A in mammals remain largely unclear.
- DNMT1
- DNMT3A
- DNMT3B
DNMT1 recognizes hemi-methylated DNA; consequently, after DNA replication and cell division, it methylates the newly synthesized strand to maintain methylation patterns of the original template stand (maintenance methylation). DNMT3A and 3B appear to primarily methylate fully unmethylated CpG sites (de novo methylation).
DNA methylation is critical for a host of biological process, including:
- X-chromosome inactivation,
- genomic imprinting,
- transcriptional silencing, and
- the maintenance of cellular identity by enabling tissue-specific gene expression.
Despite the majority of DNA methylation being found at CpG dinucleotides, the presence of non-CG methylation within mammals has now also been described. This appears localized to CpH sites (where H = A, C, or T), and is highly enriched in neurons, where it is deposited during synaptogenesis. CH methylation is also found at lower density in a range of stem cells, as well as blood and umbilical cord in a tissue-specific manner, though much remains unclear as to its regulation and potential role in development and disease.
2. Histone Variants and Histone Modifications
Nucleosomes: DNA is tightly woven around histone proteins, forming compact complexes of DNA and protein called ‘nucleosomes.’ Nucleosomes, in turn, are packed together to form chromatin (see Figure 1). Within the nucleosomes, histone proteins are arranged in an eight-part configuration, consisting of two copies of each of histones H2A, H2B, H3, and H4.
Histone variants: In addition to these canonical histones, there are numerous histone variants that show tissue and/or developmental specificity of expression, independent of DNA replication. For instance, H3 has at least three variants, H3.1, H3.2, and H3.3, which differ by only a few amino acids. Nevertheless, they have very different expression patterns and functions. This is similarly the case for variants of other histone proteins.
Additionally, some histone variants show a restricted chromosomal location associated with their function, including centromere-specific H3 variant histone (such as CENP-A), located to a specific region on every chromosome and essential for chromosome segregation.
Histone modifications: Some histones contain long terminal ‘tails’ (particularly on the N-terminus) that sit ’outside’ of the core nucleosomal structure and are therefore accessible to a range of modifying proteins. These tails can be covalently modified at many positions in a variety of ways by the addition of specific molecular groups, including acetyl, methyl, phospho, ubiquitin, and other molecules. These are called ‘post-translational histone modifications’ or ‘histone marks’ and collectively are known as ‘histone code’ (see Figure 1).
Histone modifications involve various posttranslational chemical alterations to the amino acids of histone tails, including:
- acetylation of lysine,
- methylation of lysine and arginine,
- phosphorylation of serine and threonine, and
- the ubiquitination of lysine.
Each type of histone mark can be added and removed by a family of proteins. For example, acetylation marks on H3 and H4 are added and removed by histone acetyl transferases (HATs) and deacetylases (HDACs), respectively, while methylation groups are added and removed by specific histone methyltransferases and demethylases.
There are various mechanisms by which chemical modification of CpG sites and histones are thought to influence gene expression. The methyl group from 5-methylcytosine may block transcription factors either directly or through the recruitment of a methyl-binding protein. Otherwise, the DNMT enzymes acting on CpG sites may be physically linked to other enzymes, which generate methylation and deacetylation.
Chromatin remodeling: Even though chromatin remodeling is intricately regulated, a simplified summary is that:
- On the one hand, the hyperacetylation of histones and hypomethylation of histones and CpGs is associated with a euchromatin (open) configuration, usually associated with facilitation of gene transcriptional activity.
- On the other hand, hypoacetylation of histones and hypermethylation of histones and CpGs is associated with a heterochromatin (closed) structure and transcriptional repression.
3. Non-coding RNAs
While noncoding RNAs do not code for proteins, many are functional and may affect gene expression (see Figure 1). Of those that influence gene expression, micro RNAs (miRNAs) have been the most studied thus far. miRNAs are short pieces of RNA (~ 22 nucleotides) that affects the epigenome through binding to target mRNAs controlling the expression of key regulators such as DMNTs and histone deacetylases. In turn, CpG methylation and histone modifications can influence the transcription of certain miRNA classes. MicroRNAs may also affect gene expression directly by binding to messenger RNAs, repressing their translation.
Time Points of Plasticity in the Epigenome
Times of increased cell turnover, such as during fetal development and infancy, may be particularly susceptible both to epigenetic errors and to environmental influences. The notion that environmental exposures during ‘critical windows’ in development can have profound effects on long-term health and disease risk into adult hood has received strong evidential support from observational and experimental research [3-6].
A focus in DOHaD (developmental origins of health and disease) research has been to investigate when during development the conceptus is most vulnerable to such adverse influences, informing targeted interventions. Research initially focused on the fetal and infant periods of life, but this was followed by the general recognition that critical windows occur more widely across development, spanning from pre-conception to adolescence. This has led to the consensus terminology of the developmental origins of health and disease (DOHaD) [3].
In this blog, we focus on DNA methylation and the in utero period, to include periconception, because this falls within the ‘first 1,000 days window,’ and is also a period of remarkably rapid cell differentiation and complex epigenetic remodeling.
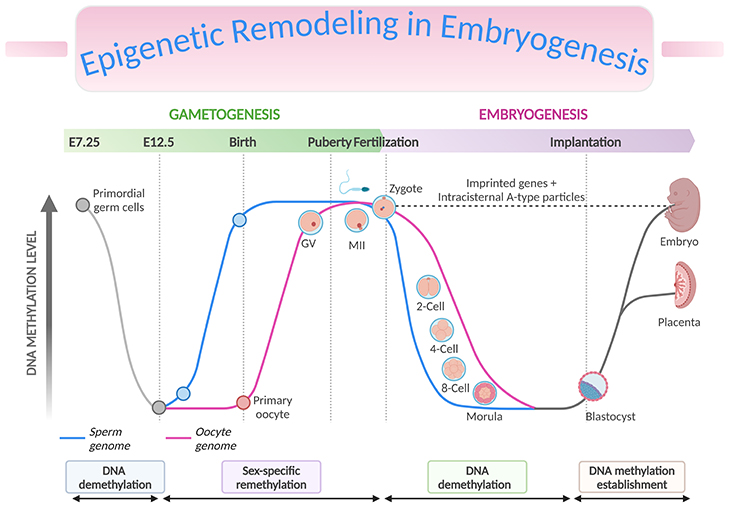
Figure 3. Epigenetic Remodeling in Embryogenesis. Genome-wide and imprint methylation programming during early development (based on mice). The time of the events depicted in the graph corresponds approximately to the events, such as methylation imprint erasure, re-establishment, and maintenance at the germline differentially methylated regions (gDMRs) during gametogenesis and early embryonic development. As primordial germ cells (PGCs) of the developing embryo enter the genital ridge, genome-wide demethylation occurs which erases the imprint marks (solid grey lines) present on the maternal and paternal chromosomes (DNA demethylation), and de novo methylation follows to establish the new sex-specific imprints during gametogenesis (sex-specific remethylation). Following fertilization, global demethylation occurs. The paternal pronucleus undergoes active demethylation which is completed before the first DNA replication while the maternal chromosome is demethylated by a DNA replication-dependent passive mechanism (DNA demethylation). The imprint marks established in the parental gametes resist the demethylation and are maintained. Genome-wide remethylation takes place around the time of implantation, and these marks are faithfully propagated to the daughter cells throughout the development of individuals (DNA methylation establishment). The inner cell mass (ICM, giving rise to the fetus) have been found to carry higher methylation than the trophectoderm (TE, giving rise to the placenta). [Intracisternal A-type particles (IAPs), IAP sequences are endogenous retrovirus-like mobile elements, present at 1000 copies in the mouse genome; GV, germinal vesicle – is the nucleus of the oocyte; MII, metaphase II stage oocytes]
During the first 48 hours after fertilization, there is rapid and active demethylation of the paternal genome and a slower and passive demethylation of the maternal genome (see Figure 3, DNA demethylation). Erasing the epigenetic marks in the zygote prior to the blastocyst stage is essential to enable pluripotency of the developing cells. Imprinted genes and some retrotransposons are known to resist demethylation at this stage. Retrotransposons are mobile elements which move in the host genome by converting their transcribed RNA into DNA through the reverse transcription. Remethylation then occurs in tissue-specific patterns after implantation, during the process of gastrulation and differentiation of the somatic cells throughout pregnancy (see Figure 3, DNA methylation establishment) [7-8].
A second wave of demethylation occurs during the epigenetic reprogramming of primordial germ cells (PGCs) in the developing embryo at the moment of their migration to the genital ridge. Parental imprints are erased at this stage in preparation for the laying down of sex-specific imprints in the PGCs. Remethylation of sperm cells occurs before the birth of the child, and in oocytes over the duration of their maturation.
The periconceptional period is therefore one of huge dynamism in the methylome, representing a window in which epigenetic errors could have significant consequences for the health of the child.
Take Home Messages
Epigenetic Layers and Players
- Epigenetic modification, including DNA methylation, histone modifications, noncoding RNA, chromatin loops, and chromatin modifying proteins provide layers of long-lasting gene regulation beyond the DNA sequence. Collectively, these can be considered an ‘epigenomic landscape’ that provides contextual information on top of existing genomic sequence maps.
Genes, Environment, and the Epigenome
- The epigenomic landscape plays a critical role during development reinforcing cell lineage commitment and migration.
- Both genetic variation and environmental factors can affect epigenetic modifications, so the epigenomic landscape reflects the complex interface of ‘nature and nurture.’
Epigenetic Regulation in Neurodevelopment
- The dynamic epigenomic landscape of the human brain is especially sensitive to environmental influences throughout both the prenatal and postnatal stages of development.
Epigenetic Dysregulation in ASD
- Epigenetic regulation may explain certain aspects of autism spectrum disorder (ASD) etiology, and early studies have shown consistent alterations in DNA methylation in both the brain and peripheral tissues.
Summary and Conclusions
Epigenetic modifications impart a layer of regulation beyond the DNA sequence and happen at the interface of genetic and environmental factors. The epigenomic landscape plays a critical role during development (i.e., throughout the span of gametogenesis, embryogenesis, and beyond) by reinforcing cell lineage commitment.
The development of nervous system is an epigenetically sensitive process due to the unique characteristics and environmental responsiveness of the neuronal epigenome. As a result, epigenetic regulation may explain certain aspects of the etiology of ASD, a complex and heterogeneous disorder with influences from genetic and environmental risk factors and with developmental origins.
Preliminary studies have shown consistent alterations in DNA methylation in both the brain and peripheral tissues, and prospective studies involving infants and children, have the potential to provide deep insight into early mechanisms and biomarkers for ASD.
Did You Know? Folate Receptor Autoantibodies (FRAAs) may impede proper folate transport.
Folate (vitamin B9) is very important for your child’s brain development!
During pregnancy, it helps prevent neural tube defects and plays a big role in forming a normal and healthy baby’s brain and spinal cord. Folate also helps cells divide and assists in both DNA and RNA synthesis.
Emerging research suggests that the presence of FRAAs negatively impacts folate transport into the brain.
- Recent studies reveal that a large subgroup of children with autism spectrum disorder (ASD) have FRAAs.
- This suggests that a possible disruption in folate transport across the blood-cerebrospinal fluid (CSF) barrier may potentially influence ASD-linked brain development.
- Screening for the FRAAs in your child should be part of your early intervention strategies.
Is there a test for identifying Folate Receptor Autoantibodies (FRAAs)?
Yes, there is a test – The Folate Receptor Antibody Test (FRAT®) has emerged as a diagnostic tool for detecting the presence of FRAAs.
It is important to screen at an early age or as soon as possible as there may be corrective measures available. Please consult your physician for further information.
To request a test kit, click on the button below.

For information on autism monitoring, screening and testing please read our blog.
References
- Mattei AL, Bailly N, Meissner A. DNA methylation: a historical perspective. Trends Genet. 2022 Jul;38(7):676-707. doi: 10.1016/j.tig.2022.03.010. Epub 2022 Apr 30. PMID: 35504755.
https://pubmed.ncbi.nlm.nih.gov/35504755/ - Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013 Jan;38(1):23-38. doi: 10.1038/npp.2012.112. Epub 2012 Jul 11. PMID: 22781841; PMCID: PMC3521964.
https://pubmed.ncbi.nlm.nih.gov/22781841/ - Hanson MA, Gluckman PD. Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev. 2014 Oct;94(4):1027-76. doi: 10.1152/physrev.00029.2013. PMID: 25287859; PMCID: PMC4187033.
https://pubmed.ncbi.nlm.nih.gov/25287859/ - Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003 Mar;33 Suppl:245-54. doi: 10.1038/ng1089. PMID: 12610534.
https://pubmed.ncbi.nlm.nih.gov/12610534/ - Law PP, Holland ML. DNA methylation at the crossroads of gene and environment interactions. Essays Biochem. 2019 Dec 20;63(6):717-726. doi: 10.1042/EBC20190031. PMID: 31782496; PMCID: PMC6923319.
https://pubmed.ncbi.nlm.nih.gov/31782496/ - Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol. 2011 Apr;31(3):363-73. doi: 10.1016/j.reprotox.2010.12.055. Epub 2011 Jan 20. PMID: 21256208; PMCID: PMC3171169.
https://pubmed.ncbi.nlm.nih.gov/21256208/ - Messerschmidt DM, Knowles BB, Solter D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014 Apr 15;28(8):812-28. doi: 10.1101/gad.234294.113. PMID: 24736841; PMCID: PMC4003274.
https://pubmed.ncbi.nlm.nih.gov/24736841/ - Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001 Jan;2(1):21-32. doi: 10.1038/35047554. PMID: 11253064.
https://pubmed.ncbi.nlm.nih.gov/11253064/


