Table of Contents
- Introduction
- Modes of Inheritance
- Phenylketonuria (PKU)
- Pathophysiology of PKU
- Biochemical Basis of the Symptoms in PKU
- Metabolic Fates of Amino Acids
- Metabolic Fate of Tyrosine
- Treatment of PKU
- Take Home Messages
- Conclusion
- Did You Know About Folate Receptor Autoantibodies (FRAAs) and Brain Development?
- References
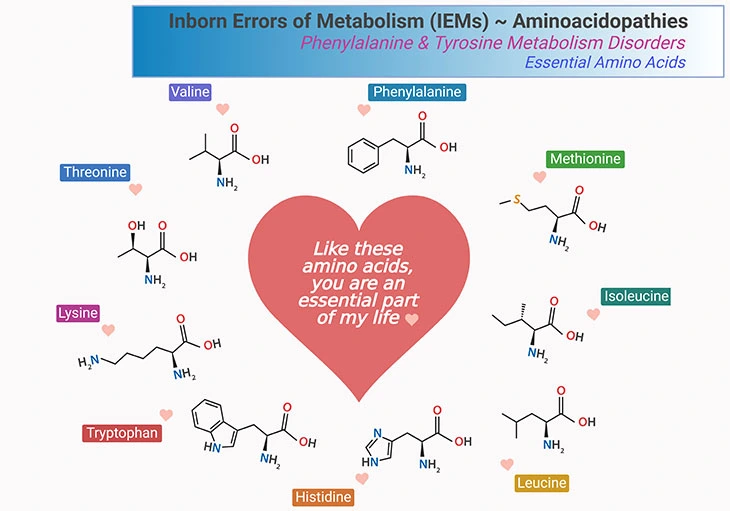
Figure 1. Essential Amino Acids. What are the metabolic fates of amino acids? Most of the amino acids have two fates: (a) incorporation into protein; and (b) conversion to various biomolecules. There are three types of amino acids: (i) essential, (ii) nonessential, and (iii) conditionally essential amino acids. As shown, there are 9 essential amino acids that human body cannot synthesize and consequently need to consume in our diet. These amino acids are phenylalanine, methionine, isoleucine, leucine, histidine, tryptophan, lysine, threonine, and valine.
Introduction

Inborn errors of metabolisms (IEMs) or inherited metabolic disorders (IMDs), are a heterogeneous group of genetic conditions that manifest mostly in childhood. Although individually rare, collectively they are numerous and cause substantial morbidity and mortality. Most having an incidence of less than 1 per 100,000 births. However, when considered collectively, the incidence may approach 1 in 800 to 1 in 2500 birth [1, 2].
Some IMDs present acutely and lead to death in the neonatal period, whereas others are benign. Some may be diagnosed in infancy, and need complex, often dietary treatment requiring lifelong monitoring. Many cause significant health problems for affected individuals.
A specific diagnosis enables genetic counselling to be offered to the family and subsequent antenatal diagnosis in future pregnancies. As girls with IMDs reach adulthood, care during pregnancy becomes an additional critical issue, especially, to achieve optimum outcome for the baby without compromising the mother’s overall health. The field of IMDs is vast and ever expanding.
Modes of Inheritance
Human beings have 23 pairs of chromosomes (Greek: Khroma ‘color’ + soma ‘body’). For each pair of chromosome, one is inherited from the father and one from the mother. One pair is the sex chromosomes: males have one X and one Y chromosome (i.e., XY), whereas females have two X chromosomes (i.e., XX). The nonsex chromosomes are called autosomes. Inherited metabolic disorders are due to mutations in the gene (DNA sequence) on the chromosomes. Autosomal recessive, autosomal dominant, and X-linked recessive are forms of Mendelian inheritance, as shown in Figure 2. (cf. previous blog entitled as ‘Developmental Delays in Infancy and Early Childhood: Branched-Chain Amino Acid and Organic Acid Metabolism Disorders’ )
(i) Autosomal Recessive Inheritance
This particular situation is shown in Figure 2A. Both parents are heterozygous, i.e., they carry an abnormal recessive gene. Carriers are usually asymptomatic, and most families are unaware that they carry a disorder until they have an affected child. For each pregnancy there is a 1 in 4 risk of the child inheriting one abnormal gene from each parent and thus being affected with the disorder. On average, 25% of offspring will be affected (abnormal homozygotes). 25% will be normal, and 50% will be carriers (heterozygotes). Significantly, most IMDs have an autosomal recessive mode of inheritance.
(ii) Autosomal Dominant Inheritance
This particular situation is illustrated in Figure 2B. An affected parent has one copy of an abnormal dominant gene (and is affected with the disorder). On average, half of that parent’s offspring will inherit the abnormal gene and will be affected with the same disorder as their parent. Half of the parent’s offspring will not inherit the gene and so they will not be affected with the disorder, nor will they be a carrier. For example, some of the porphyrias are inherited in an autosomal dominant fashion.
(iii) X-Linked Inheritance
X-linked disorders are due to mutations on the X chromosome. Males inherit their one X chromosome from their mother. If it carries a mutation they will be affected with the corresponding disorder, i.e., they will be a hemizygote. This particular situation is shown in Figure 2C.
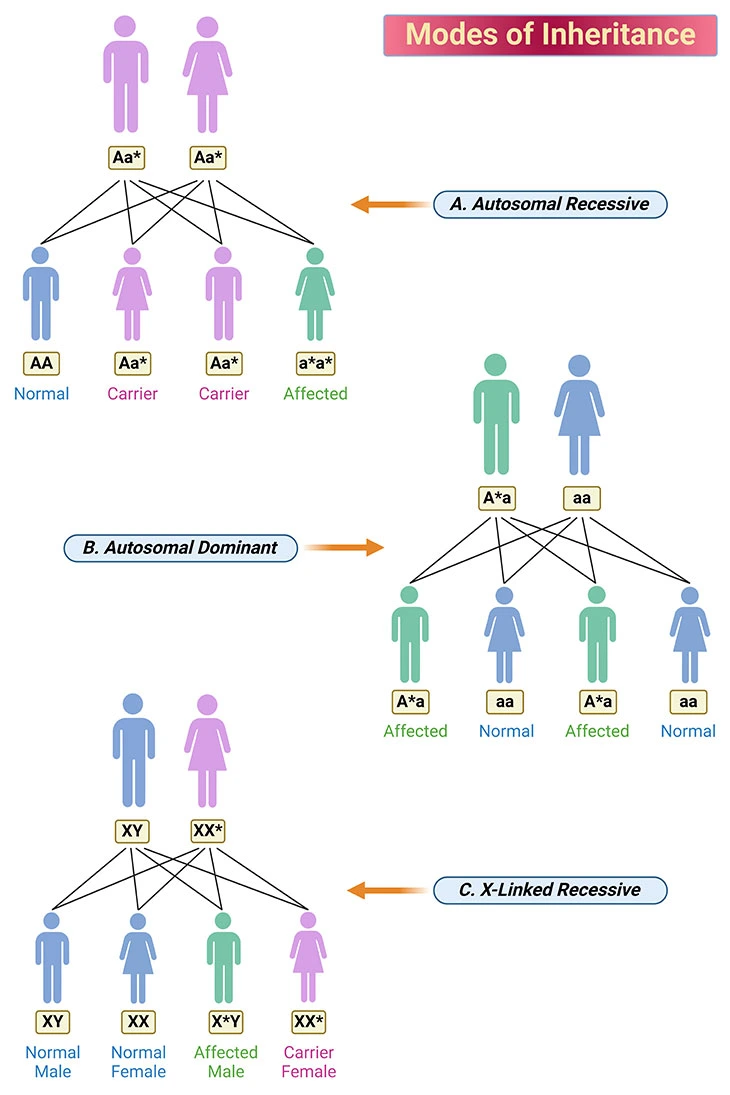
Figure 2. Modes of Inheritance. The modes of inheritance illustrated are: (A) autosomal recessive; (B) autosomal dominant; and (C) X-linked recessive. The chromosomes carrying a mutation is indicated with an asterisk (*). A blue individual is normal, a purple individual is carrier, and a green individual is affected with the disorder. {A, dominant gene on autosome; a, recessive gene on autosome; *, disease causing gene.}
In females, a process called ‘lyonization’ randomly inactivates one of the X chromosomes in each cell of the body. Consequently, in some female heterozygotes there is a chance that more abnormal than normal X chromosomes will be active. This can result in a female having symptoms of the disorder. If affected, females generally have a milder form the disorder, but nevertheless they may have significant clinical problems. Examples of X-linked disorders are adrenoleukodystrophy and the urea cycle disorder ornithine transcarbamylase deficiency.
Phenylketonuria (PKU)

PKU is a disorder of amino acid metabolism that causes a clinical syndrome of intellectual disability with cognitive and behavioral abnormalities caused by elevated serum phenylalanine. The primary cause is deficient phenylalanine activity. Diagnosis is by detecting high phenylalanine levels and normal or low tyrosine levels. Treatment is lifelong dietary phenylalanine restriction. Prognosis is excellent with treatment
(see Box – 1) [1, 2].
The majority of PKU cases are referred to as ‘classic ’ and involve a deficiency of the phenylalanine hydroxylase enzyme (PAH), a small number have an elevated phenylalanine level due to a defect in dihydrobiopterin reductase enzyme (DHPR) activity (nonclassical PKU) that regenerates tetrahydrobiopterin (BH4) cofactor for PAH enzyme activity (see Figure 3, 4).
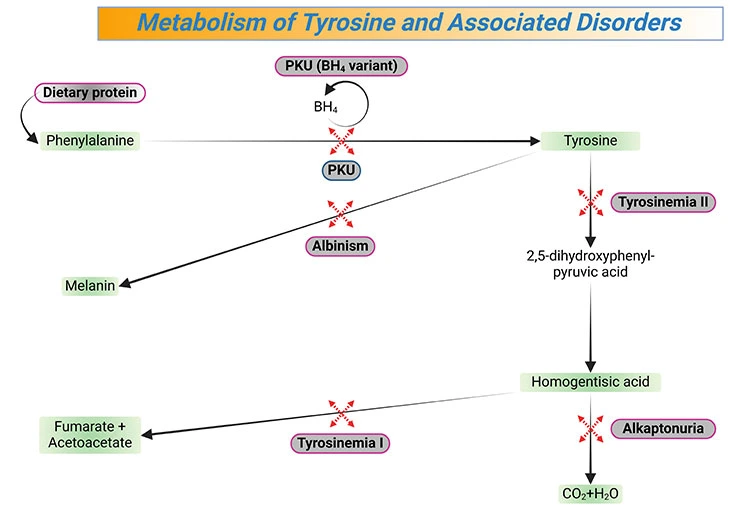
Figure 3. Metabolism of Tyrosine and Associated Disorders. Tyrosine is an amino acid that is a precursor of several neurotransmitters (e.g., dopamine, norepinephrine, epinephrine), hormones (e.g., thyroxine), and melanin; deficiencies of enzymes involved in its metabolism lead to a variety of syndromes, (e.g., tyrosinemia I, tyrosinemia II, alkaptonuria, albinism). [PKU, classical phenylketonuria; PKU (BH4 variant), nonclassical phenylketonuria]
The newborn with PKU may appear functionally normal, early detection through newborn screening identifies PKU at a time when early treatment can prevent development of the cardinal manifestation, viz., intellectual impairment. Should this escape detection or not be performed, then the affected individual develops a peculiar ‘mousy odor’ particularly in the urine, very fair complexion, hair, and pale eye color. This is in fact related to impairment of melanin synthesis. Additionally, child may exhibit missed developmental milestones such as walking and talking and later on will manifest severe intellectual impairment, eczema, seizure activity, and self-abusive behavior unless treatment is initiated promptly [2, 4, 5].
Pathophysiology of PKU
When an excessive amount of phenylalanine crosses the blood-brain barrier (BBB), is toxic to the myelin sheath. Moreover, a decrease in the biosynthesis of dopamine, epinephrine, and norepinephrine explains the neurologic dysfunctions. Unless this is aggressively approached with dietary manipulation, irreversible damage occurs before 6 months of age. Besides, the low tyrosine level, due to the inability to convert phenylalanine to tyrosine, results in failure of pigment formation (pseudoalbinism) and consequently, would explain the light-colored eyes and skin (see Figure 3).
A normal nutritional diet contains an excess of phenylalanine necessary for normal metabolic requirements. Conversion to tyrosine occurs in the liver. Hundreds of mutations in the phenylalanine hydroxylase gene have been defined with impairment ranging from 50% activity to total enzyme inactivity. Only minor reduction in the enzyme activity may result in elevated levels of phenylalanine without neurological impairment (hyperphenylalaninemia). When the enzyme deficiency reaches critical levels, phenylalanine excess is ‘shunted’ into alternative metabolic pathways. It is these metabolic products, viz., phenylpyruvate and phenylacetate that are responsible for the odor in the urine of the infant. Strict dietary management of PKU can achieve a near normal phenylalanine level and, in turn, normal development.
For this above reason, mandatory screening of all newborn infants has become the standard of care. Most importantly, PAH-deficient expectant mothers must be judicious in their dietary restriction of phenylalanine before and during pregnancy. Elevated levels of phenylalanine result in serious teratogenic damage in utero i.e., toxic embryopathy, resulting in microcephaly and cardiac abnormalities in this situation.
Biochemical Basis of the Symptoms in PKU
PKU falls into the aminoacidopathies category of IEMs, due to the defective metabolism of amino acids (see Figure 1). Examples of various diseases that are associated with phenylalanine and tyrosine metabolism are illustrated in Figure 3 and 4. Accumulating molecules may be toxic giving rise to symptoms such as hyperactivity, intellectual disability, developmental delay, vomiting, and seizures [3, 4].
Metabolic Fates of Amino Acids
Fundamentally, most of the amino acids have two metabolic fates: viz., (a) incorporation into protein; and (b) conversion to various biomolecules. There are 3 types of amino acids: i.e., (i) essential; (ii) nonessential, and (iii) conditionally essential amino acids (see Table – 1; Figure 1).

Metabolic Fate of Tyrosine
Tyrosine is synthesized from phenylalanine by phenylalanine hydroxylase using tetrahydrobiopterin (BH4) as a cofactor. In this context, tyrosine is the precursor of dopamine, norepinephrine, and epinephrine, also known as catecholamine neurotransmitters. It is also the precursor of the thyroid hormones and melanin. The cofactor tetrahydrobiopterin is essential not only for tyrosine synthesis but also for the synthesis of L-DOPA, the precursor for the catecholamines and for the synthesis of serotonin from tryptophan. These reactions are hydroxylations by monooxygenases using tetrahydrobiopterin as a cofactor. During this reaction, tetrahydrobiopterin is oxidized to dihydrobiopterin (BH2) Recycling of dihydrobiopterin back to tetrahydrobiopterin by dihydropteridine reductase (DHPR) is required for the continued synthesis of tyrosine, L-DOPA, and serotonin (see Figure 4).
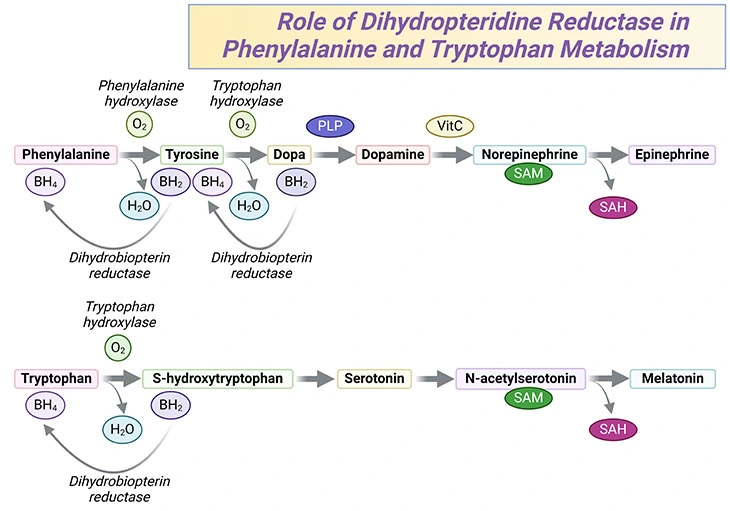
Figure 4. Phenylalanine and Tryptophan Metabolism. The role of dihydropteridine reductase in phenylalanine and tryptophan metabolism in the synthesis of neurotransmitters, such as dopamine, norepinephrine, serotonin, as well as hormones such as epinephrine and melatonin.
Hyperphenylalaninemia may arise due to a deficiency in either phenylalanine hydroxylase or DHPR (see Figure 3 and 4) Both of these deficiencies are considered as phenylketonuria. However, since tetrahydrobiopterin is a cofactor in several reactions, the effects of DHPR deficiency may cause even more severe neurological difficulties than those with phenylalanine hydroxylase deficiency. Patients with DHPR deficiency may also be less response to therapy.
Failure to convert phenylalanine to tyrosine results in the accumulation of phenylalanine, which becomes a major donor of amino groups in aminotransferase. As a result, phenylpyruvate is synthesized. The urinary phenylpyruvate accumulation is used as a diagnostic tool for PKU. Phenylpyruvate is further oxidized to phenylacetate, which imparts a ‘mousy’ odor to the urine.
The progressive mental impairment in untreated PKU is due to several reasons. For example:
- depletion of tyrosine as discussed earlier;
- deletion of neural tissue α-ketoglutarate, which is essential for the energy production from the TCA cycle; and
- depletion of other large neutral amino acids, which use the same amino acid transporters to cross the blood-brain barrier as phenylalanine. High levels of phenylalanine can competitively block the uptake of amino acids by the brain that are required for protein and neurotransmitter synthesis.
Treatment of PKU
- Dietary phenylalanine restriction is critical. A strict dietary regimen is required to control the devastating effect of this defect on cognitive function.
- Treatment relies on careful control of intake of phenylalanine that is sufficient for protein synthesis and yet drastically reduces the level of phenylalanine in the diet to prevent its accumulation.
- Immediate dietary therapy is enacted with a combination of a special formula lacking phenylalanine and a small amount of natural protein to provide sufficient, but not excess, phenylalanine and tyrosine for growth and development.
- Phenylalanine levels drop to normal, and symptoms improve. If treatment is initiated before 3 weeks of life, intellectual development is near normal. However, the longer treatment is delayed, the worse the cognitive outcome.
- Ideally, dietary restrictions should be maintained throughout life. This, as one might easily imagine, presents a formidable challenge for the patient [5].
Take Home Messages
- Phenylalanine hydroxylase (PAH) deficiency is responsible for classic phenylketonuria (PKU).
- Tetrahydrobiopterin (BH4) is an essential cofactor in phenylalanine metabolism. In a small percentage of PKU cases, the defect is in BH4 metabolism (and not PAH) that must be corrected to prevent neurological damage.
- PKU is caused by one of several gene mutations that result in deficiency or absence of phenylalanine hydroxylase so that dietary phenylalanine accumulates; the brain is the main organ affected, possibly because of disturbance of myelination.
- PKU causes a clinical syndrome of intellectual disability with cognitive and behavioral abnormalities; if untreated, the intellectual disability is severe.
- In the US and many developed countries, all neonates are screened for PKU 24 to 48 hours after birth with one of several blood tests; abnormal results are confirmed by directly measuring phenylalanine levels.
- Treatment is lifelong dietary phenylalanine restriction; adequate treatment begun in the first days of life prevents many manifestations of the disease.
- Although prognosis is excellent with treatment, frequent monitoring of plasma phenylalanine levels is required; recommended targets are between 2 mg/dL and
6 mg/dL (129 to 360 μmol/L) for all children. - Toxic phenylalanine metabolites in the mother’s circulation can lead to damage in utero if the PKU pregnant woman is not compliant with the diet prior to conception through term.
- Tyrosine is an amino acid that is a precursor of several neurotransmitters (e.g., dopamine, norepinephrine, epinephrine), hormones (e.g., thyroxine), and melanin; deficiencies of enzymes involved in its metabolism lead to a variety of syndromes.
Conclusion
In this blog, we presented an overview of numerous disorders of phenylalanine and tyrosine metabolism. PKU, a rare, inherited defect in the metabolism of the amino acid phenylalanine, was first described in 1934. Consequently, the discovery of PKU was considered as a fundamentally important discovery for the fields of biochemistry, genetics, metabolism, and medicine. The creation and implementation of newborn screening programs for PKU and other rare inherited metabolic disorders, and the subsequent, successful treatment of affected infants, is a huge success story!
Did You Know? Folate Receptor Autoantibodies (FRAAs) may impede proper folate transport.
Folate (vitamin B9) is very important for your child’s brain development!
During pregnancy, it helps prevent neural tube defects and plays a big role in forming a normal and healthy baby’s brain and spinal cord. Folate also helps cells divide and assists in both DNA and RNA synthesis.
Emerging research suggests that the presence of FRAAs negatively impacts folate transport into the brain.
- Recent studies reveal that a large subgroup of children with autism spectrum disorder (ASD) have FRAAs.
- This suggests that a possible disruption in folate transport across the blood-cerebrospinal fluid (CSF) barrier may potentially influence ASD-linked brain development.
- Screening for the FRAAs in your child should be part of your early intervention strategies.
Is there a test for identifying Folate Receptor Autoantibodies (FRAAs)?
Yes, there is a test – The Folate Receptor Antibody Test (FRAT®) has emerged as a diagnostic tool for detecting the presence of FRAAs.
It is important to screen at an early age or as soon as possible as there may be corrective measures available. Please consult your physician for further information.
To request a test kit, click on the button below.

For information on autism monitoring, screening and testing please read our blog.
References
- Gonzalez J and Willis MS. Ivar Asbjörn Følling: Discovered Phenylketonuria (PKU). Laboratory Medicine, Volume 41, Issue 2, February 2010, Pages 118–119.
https://doi.org/10.1309/LM62LVV5OSLUJOQF - Kruszka P, Regier D. Inborn Errors of Metabolism: From Preconception to Adulthood. Am Fam Physician. 2019 Jan 1;99(1):25-32. PMID: 30600976.
https://pubmed.ncbi.nlm.nih.gov/30600976/ - McDonald L, Rennie A, Tolmie J, Galloway P, McWilliam R. Investigation of global developmental delay. Arch Dis Child. 2006 Aug;91(8):701-5. doi: 10.1136/adc.2005.078147. PMID: 16861488; PMCID: PMC2083045.
https://pubmed.ncbi.nlm.nih.gov/16861488/ - Williams RA, Mamotte CD, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008 Feb;29(1):31-41. PMID: 18566668; PMCID: PMC2423317.
https://pubmed.ncbi.nlm.nih.gov/18566668/ - Vockley J, Andersson HC, Antshel KM, Braverman NE, Burton BK, Frazier DM, Mitchell J, Smith WE, Thompson BH, Berry SA; American College of Medical Genetics and Genomics Therapeutics Committee. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med. 2014 Feb;16(2):188-200. PMID: 24385074.
https://pubmed.ncbi.nlm.nih.gov/24385074/


