Table of Contents
- Introduction
- Biological Methylation: The Folate Connection
- DNA Methylation and Cancer
- Aberrant Methylation in Carcinogenesis
- Effects of Folate and One-Carbon Nutrients on DNA Methylation
- Human Evidence: Folate Status and Genomic DNA Methylation
- Gene-Specific Methylation: Folate’s Dual Role in Carcinogenesis
- Take-Home Messages
- Summary and Conclusions
- Did You Know About Folate Receptor Autoantibodies (FRAAs) and Brain Development?
- References

Figure 1. The Dual Edge of Folate: DNA Health, Supplementation, and Autism Care. This infographic illustrates folate’s paradoxical role in health, showing how its benefits and risks depend on dose, genetic background, and clinical context. On the left, Essential for DNA Health highlights folate’s central role in one-carbon metabolism, supporting DNA methylation, repair, and genomic stability. Adequate folate intake prevents mutations and maintains healthy cell division, while deficiency is linked to hypomethylation, chromosomal instability, and increased cancer risk. Emerging DNA methylation biomarkers underscore its importance as a guardian of genomic integrity. In the center, Complexity of Supplementation emphasizes that while folate supplementation can correct deficiencies, excess intake risks hypermethylation and inappropriate gene silencing. Genetic subgroups, such as individuals with MTHFR polymorphisms, may require tailored doses, and safe thresholds remain under investigation. This reflects the delicate balance between protection and disruption, reminding us that “more” is not always better. On the right, Implications for Autism Care show how folinic acid and high folate intake are sometimes used to support neurodevelopment in children with autism. Yet supraphysiological dosing may alter methylation patterns, raising concerns about long-term cancer risk. Precision in dosing is therefore crucial, especially in pediatric populations, to balance therapeutic promise against unintended genomic consequences. Together, the figure conveys folate’s dual nature—as both protector and potential disruptor—underscoring the need for individualized, context-specific approaches to supplementation and therapy.
Introduction
Folate, Methylation, and the Delicate Balance of Genomic Integrity
Folate and its derivatives occupy a central role in one-carbon metabolism, serving as indispensable cofactors for the synthesis of S-adenosylmethionine (SAM), the universal methyl donor. Through this pathway, folate orchestrates the methylation of DNA, RNA, proteins, and phospholipids, thereby safeguarding genomic integrity, regulating gene expression, and maintaining cellular homeostasis. Yet, as our discussion has underscored, the relationship between folate and methylation is not linear but bidirectional and context-dependent. Both deficiency and excess supplementation can perturb methylation patterns, producing either global hypomethylation or gene-specific hypermethylation, each with profound implications for carcinogenesis [1 – 9].
The dual effect paradigm—where folate acts as both protector and potential disruptor—has emerged from decades of experimental and clinical studies. Genomic hypomethylation, often observed in folate deficiency, destabilizes chromosomal architecture and predisposes to recombination events. Conversely, promoter hypermethylation of tumor suppressor genes under conditions of altered folate metabolism can silence critical pathways of cellular defense. This paradox mirrors the methylation abnormalities seen in epithelial cancers, lending credence to the hypothesis that folate status is a determinant of cancer risk [1 – 9].
Within this framework, a contemporary concern has surfaced: whether indiscriminate consumption of excess folinic acid—particularly in vulnerable populations such as children with autism spectrum disorder (ASD)—might inadvertently increase long-term cancer risk. Folinic acid (5-formyltetrahydrofolate) is widely used in clinical practice, including as an adjunct in ASD management, where it has shown promise in improving language and behavioral outcomes in subsets of children with folate receptor autoantibodies. However, scientific literature cautions that supraphysiological folate exposure may alter DNA methylation dynamics, potentially fostering site-specific hypermethylation of tumor suppressor genes or enhancing susceptibility to C→T transition mutations in hypermutable regions such as p53 exons 5–8. While direct evidence linking folinic acid supplementation in autistic children to carcinogenesis is currently lacking, the broader body of research on folate’s dual effects in cancer biology suggests that dose, duration, genetic background (e.g., MTHFR polymorphisms), and tissue environment are critical determinants of outcome (see Figure 1) [10].
Thus, the challenge is not simply to recognize folate as a nutrient essential for health, but to appreciate its nuanced role as a double-edged sword. The following synthesis explores how folate and other one-carbon nutrients modulate DNA methylation at both genomic and gene-specific levels, highlighting the delicate balance between protection and risk, and underscoring why precision in supplementation—rather than indiscriminate use—is vital for safeguarding long-term genomic stability.
I. Biological Methylation: The Folate Connection
S-Adenosylmethionine as the Universal Methyl Donor
In mammalian cells, most methylation reactions rely on S-adenosylmethionine (S-AdoMet or SAM) as the principal methyl donor. The labile methyl group of SAM originates from methionine, which itself acquires this moiety from 5-methyltetrahydrofolate (5-methyl-THF)—a critical folate derivative essential for the de novo synthesis of methionine. Each SAM-mediated methylation reaction yields two products: the methylated target molecule and S-adenosylhomocysteine (S-AdoHcy or SAH). Importantly, SAH is a potent inhibitor of nearly all SAM-dependent methyltransferases, with Ki values ranging between 2–6 μM, underscoring its regulatory role in cellular methylation capacity (see Figure 2).
Targets of Methylation and Folate Deficiency
The reach of SAM-dependent methylation extends across DNA, RNA, proteins, and phospholipids (see Figure 2). Experimental studies consistently demonstrate that inadequate one-carbon nutrient supply reduces methylation levels in each of these biomolecules. Among them, DNA methylation abnormalities have attracted the greatest attention, particularly in the context of carcinogenesis, where folate status profoundly influences methylation dynamics. By contrast, the potential pro-transformational effects of aberrant methylation in proteins, RNA, and phospholipids remain relatively underexplored, with only limited studies addressing these pathways.
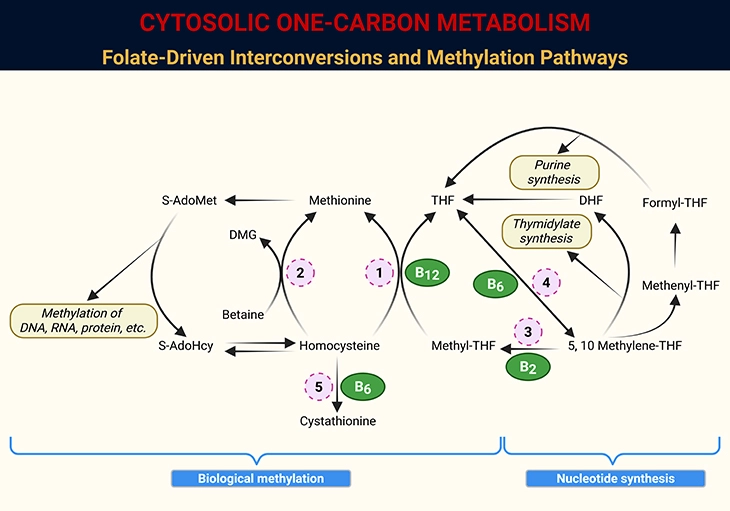
Figure 2. Cytosolic One-Carbon Metabolism: Folate-Driven Interconversions and Methylation Pathways. Schematic representation of one-carbon metabolism within the cytosol of mammalian cells, highlighting the dynamic interconversion of folate coenzymes and their roles in nucleotide synthesis and methylation. Key intermediates include tetrahydrofolate (THF), dihydrofolate (DHF), S-adenosylmethionine (S-AdoMet), S-adenosylhomocysteine (S-AdoHcy), and dimethylglycine (DMG). Enzymatic steps are catalyzed by: 1. Methionine synthase (MS); 2. Betaine-homocysteine methyltransferase (BHMT); 3. Methylenetetrahydrofolate reductase (MTHFR); 4. Serine hydroxymethyltransferase (SHMT); 5. Cystathionine β-synthase (CBS).
II. DNA Methylation and Cancer
CpG Sites and DNA Methyltransferases
In mammalian genomes, DNA methylation occurs almost exclusively at the 5-carbon of cytosine residues preceding guanine—known as CpG sites. Approximately 50–70% of CpG residues in the adult human genome are methylated. These sites cluster in CpG islands, typically located in the 5′-untranslated regions (UTRs) of genes. Under basal conditions, CpG islands remain largely unmethylated, whereas dispersed CpG sites within coding regions are heavily methylated.
The conversion of cytosine to 5-methylcytosine is catalyzed by DNA methyltransferases (DNMTs) (see Figure 3).
- DNMT1 functions as a maintenance methyltransferase, copying parental methylation patterns onto daughter strands during replication.
- DNMT3a and DNMT3b mediate de novo methylation, most prominent during embryogenesis and in cancer cells.
- DNMT2, though identified, remains enigmatic—its primary role may extend beyond DNA to RNA methylation.
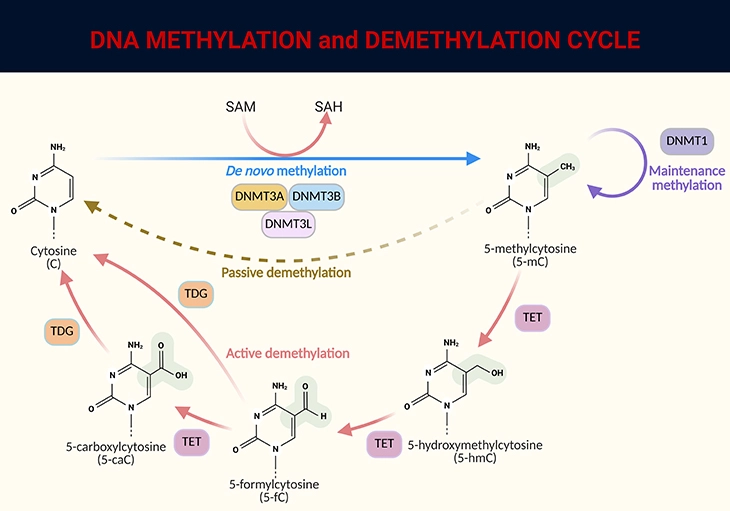
Figure 3. DNA methylation and demethylation cycle. DNA methylation involves the addition of a methyl group (CH₃) from the universal donor S-adenosylmethionine (SAM) to the fifth carbon of cytosine (C), facilitated by DNA methyltransferase (DNMT), resulting in 5-methylcytosine (5-mC). This process produces S-adenosylhomocysteine (SAH) as a byproduct. DNA demethylation occurs through passive or active processes: (1) Passive demethylation – Happens over successive cell divisions without the addition of new methyl groups. (2) Active demethylation – Involves the sequential conversion of 5-mC to 5-hydroxymethylcytosine (5-hmC), then to derivatives like 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC), ultimately returning to cytosine (C). This process is regulated by the ten-eleven translocation (TET) family of proteins. The combined actions of these mechanisms ensure that DNA methylation and demethylation are dynamic processes, crucial for regulating gene expression and maintaining cellular function.
Functional Significance of CpG Methylation
The precision with which cells preserve CpG methylation patterns highlights their importance. Some patterns are established during embryogenesis or early postnatal life, while others act as lifelong adaptive mechanisms, enabling organisms to respond to environmental cues and maintain homeostasis. CpG islands in the 5′-UTR are particularly critical, regulating gene expression: demethylation often enhances transcription, while methylation suppresses it (see Figure 4 and Figure 5).
Beyond transcriptional control, DNA methylation safeguards genetic integrity, influences chromatin organization, and modulates mutational susceptibility. For example, in p53, sites prone to C→T transition mutations frequently overlap with methylcytosine residues—a concordance strongly suggestive of causal linkage, though the precise mechanism remains debated.
III. Aberrant Methylation in Carcinogenesis
The Paradox of Hypomethylation and Hypermethylation
Epithelial cancers often exhibit two paradoxical methylation abnormalities:
- Global genomic hypomethylation
- Localized CpG island hypermethylation
The reasons why certain CpG islands are particularly vulnerable to hypermethylation in neoplasia remain unclear. However, genomic hypomethylation is consistently observed in dysplastic (premalignant) tissues and is considered one of the earliest molecular events in carcinogenesis. Studies reveal that the degree of genomic methylation decreases incrementally with advancing grades of neoplasia, reinforcing its role in tumor progression (see Figure 4 and Figure 5).
Gene-Specific vs. Genomic Effects
Cancer evolution is often attributed to gene-specific methylation changes rather than global phenomena. Indeed, hypermethylation of promoter CpG islands in tumor suppressor genes—with consequent gene silencing—is a hallmark of many cancers. Conversely, hypomethylation at critical sites may disrupt imprinting, abolishing the suppressed expression of one allele and promoting dedifferentiation and tumorigenesis, as demonstrated in colorectal cancer models [1 – 6].
Yet, genomic hypomethylation itself may be causal. By destabilizing DNA, it increases susceptibility to mitotic recombination, facilitating chromosomal breaks, translocations, and allelic loss—all of which accelerate malignant transformation (see Figure 5).
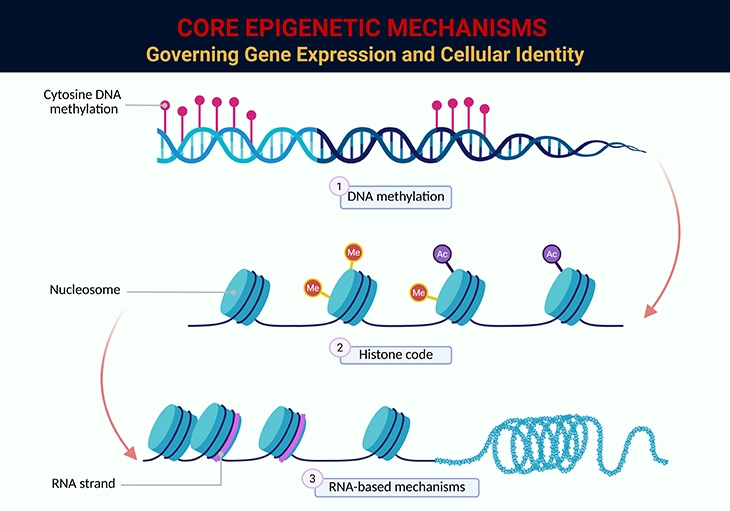
Figure 4. Core Epigenetic Mechanisms Governing Gene Expression and Cellular Identity. This figure illustrates the three principal epigenetic pathways that regulate gene expression without altering the underlying DNA sequence: 1. DNA Methylation: The addition of methyl groups (CH₃) to cytosine residues within CpG dinucleotides, typically leading to transcriptional repression. Methylation patterns are essential for genomic imprinting, X-chromosome inactivation, and silencing of repetitive elements. Aberrant methylation—either global hypomethylation or promoter hypermethylation—is implicated in carcinogenesis and developmental disorders. 2. Histone Modifications (Histone Code): Post-translational modifications of histone tails—including methylation, acetylation, phosphorylation, and ubiquitination—alter chromatin structure and accessibility. Histone acetylation generally promotes transcriptional activation by loosening chromatin, while histone methylation can either activate or repress gene expression depending on the specific residue and context (e.g., H3K4me3 vs. H3K27me3). These modifications form a dynamic “histone code” interpreted by reader proteins to orchestrate cell-type–specific transcriptional programs. 3. RNA-Based Mechanisms: Non-coding RNAs—including microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and small interfering RNAs (siRNAs)—modulate gene expression post-transcriptionally and guide chromatin remodeling complexes to specific genomic loci. These RNA species play critical roles in development, immune regulation, and disease pathogenesis, and are increasingly recognized as key mediators of epigenetic inheritance. Together, these mechanisms form a multilayered regulatory network that governs cellular identity, developmental plasticity, and disease susceptibility. Their interplay is especially relevant in contexts such as cancer, neurodevelopmental disorders, and aging, where epigenetic dysregulation can have profound consequences.
IV. Effects of Folate and One-Carbon Nutrients on DNA Methylation
Genomic Methylation: Patterns and Paradoxes
Depletion of folate—whether in cultured cells, intact animals, or humans—has been shown to induce genomic DNA hypomethylation under certain experimental conditions. Intriguingly, it may also trigger site-specific hypermethylation, a dual pattern that mirrors the abnormalities observed in epithelial cancers [1 -6]. This resemblance strengthens the hypothesis that inadequate folate availability promotes carcinogenesis through disruptions in DNA methylation. Yet, as in cancer biology, the precise mechanisms driving the coexistence of global hypomethylation and localized hypermethylation remain poorly defined.
Inconsistencies Across Studies
Despite compelling evidence, changes in DNA methylation following folate depletion have not been consistently reproducible. Variability may stem from differences in methodologies used to measure methylation, or from confounding factors such as age, which itself influences methylation status. Moreover, methylation changes are dynamic, fluctuating during the course of nutrient depletion. For example, serial studies in rodents fed a methyl-deficient diet (lacking choline, B12, and folate) revealed an initial wave of genomic hypomethylation that intensified over time. This was accompanied by an up-regulation of DNA methyltransferase activity, possibly as a compensatory response. Subsequently, site-specific hypermethylation emerged at loci receptive to enhanced methyltransferase activity. Such temporal dynamics may explain why some investigators report no change, or even paradoxical increases, in genomic methylation under folate deprivation.
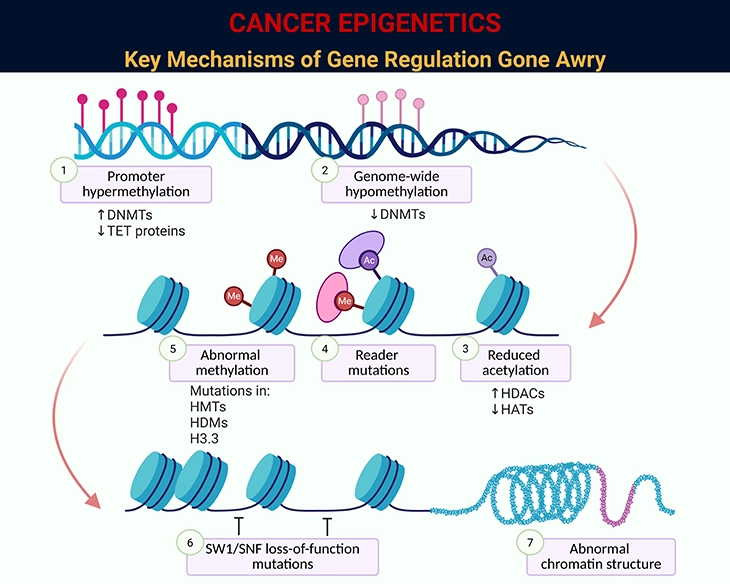
Figure 5. Cancer Epigenetics: Key Mechanisms of Gene Regulation Gone Awry. This figure illustrates the major epigenetic disruptions that contribute to cancer development. Unlike genetic mutations, these changes do not alter the DNA sequence itself but instead reshape how genes are switched “on” or “off” and how the genome is packaged and interpreted. Together, they destabilize cellular identity and promote tumor growth: 1. Promoter Hypermethylation – Excess methylation at gene start sites silences tumor-suppressor genes. For example: p16INK4A and hMLH1 are frequently hypermethylated in colorectal and gastric cancers, blocking cell-cycle control and DNA repair. 2. Genome-Wide Hypomethylation – Loss of methylation across the genome destabilizes DNA and activates harmful elements. For example: Hypomethylation of LINE-1 repetitive elements in lung and bladder cancers leads to chromosomal instability. 3. Reduced Histone Acetylation – Lower acetylation tightens chromatin, restricting access to protective genes. For example: Reduced histone H3 acetylation in breast cancer cells correlates with silencing of estrogen receptor target genes. 4. Reader Mutations – Mutations in proteins that “read” epigenetic marks misdirect gene regulation. For example: BRD4 mutations alter enhancer activity in leukemia, driving uncontrolled cell proliferation. 5. Abnormal Methylation Enzyme Mutations – Mutations in enzymes that add or remove methyl groups disrupt normal patterns. For example: DNMT3A mutations in acute myeloid leukemia (AML) cause widespread abnormal methylation and poor prognosis. 6. SWI/SNF Complex Loss-of-Function – This chromatin remodeling complex normally opens DNA for transcription; its loss impairs gene regulation. For example: SMARCB1 mutations are hallmarks of malignant rhabdoid tumors. 7. Abnormal Chromatin Structure – Altered folding of DNA changes accessibility of key genes. For example: In prostate cancer, abnormal chromatin looping brings oncogenes under strong enhancers, boosting their activity. Together, these epigenetic mechanisms silence protective pathways, destabilize the genome, and activate oncogenic programs—laying the foundation for cancer initiation and progression.
Biochemical Determinants: SAM, SAH, and the Ratio That Matters
Mechanistically, genomic hypomethylation induced by folate depletion appears to be driven primarily by increased concentrations of S-adenosylhomocysteine (S-AdoHcy), a potent methyltransferase inhibitor. In contrast, limitations in S-adenosylmethionine (S-AdoMet or SAM) availability play a lesser role. This is supported by animal models with genetically engineered defects in SAM synthesis, where methylation changes were not solely explained by SAM depletion.
The SAM/SAH ratio is often considered a proxy for the methylation potential of a biological system. However, its utility lies mainly in reflecting S-AdoHcy accumulation, since SAM levels are typically maintained within a narrow range by feedback regulation involving methylene-THF reductase (MTHFR). Importantly, the hydrolysis of SAH to homocysteine is thermodynamically unfavorable, favoring SAH accumulation and amplifying its inhibitory effect (see Figure 2).
Experimental data illustrate this paradox: in rats with severe folate deficiency, colonic mucosal folate concentrations decreased by nearly 100-fold, while colonic SAM levels increased sixfold. Yet, mucosal SAM concentrations remained unchanged, and DNA methylation did not correlate predictably with SAM levels. Similar findings have been reported in cell culture, underscoring that SAM alone is a poor predictor of genomic methylation. In sum, while SAM and SAH are partial determinants, their concentrations correlate with DNA methylation in an unpredictable fashion, reflecting the influence of additional regulatory factors [7 – 9].
Preclinical and Clinical Evidence: Magnitude, Duration, and Tissue Specificity
On a genomic level, hypomethylation induced by one-carbon nutrient depletion has been documented in both animal models and human studies. However, its occurrence depends on the severity and duration of depletion, and whether multiple nutrients are simultaneously deficient. Tissue specificity further complicates interpretation.
Comparative rodent studies highlight this variability: one experiment inducing severe folate deficiency readily demonstrated genomic hypomethylation, whereas a parallel study with moderate depletion showed no evidence of demethylation. Typically, genomic hypomethylation emerges within weeks, though the precise onset remains undefined.
The Colonic Mucosa: A Resistant Yet Vulnerable Tissue
The colonic mucosa exemplifies tissue-specific responses. Despite observations in cultured human colonocytes, intact animal studies consistently show that the colonic mucosa is highly resistant to hypomethylation under isolated folate depletion.
Yet, two factors can sensitize it:
- Synergistic depletion of multiple one-carbon nutrients. In mice, mild combined depletion of folate, B12, B6, and B2 reduced colonic genomic methylation by ~50%, even though folate depletion alone had no effect.
- Age-related genomic hypomethylation, observed across diverse tissues, may predispose the colon to dietary folate modulation. However, this sensitizing effect is not universally observed, highlighting the complexity of age–nutrient interactions.
V. Human Evidence: Folate Status and Genomic DNA Methylation
Controlled Studies and Genetic Interactions
Sustained dietary folate inadequacy has been shown to produce genomic DNA hypomethylation in humans, though the number of intervention studies remains limited. The most convincing evidence comes from highly controlled metabolic unit studies, where subjects either resided within the unit or obtained all meals under strict supervision. In these settings, white blood cell DNA hypomethylation was observed when mean plasma folate concentrations fell to 9.0–13.7 nmol/L (≈4.1–6.3 ng/mL)—a relatively modest systemic depletion.
Cross-sectional studies in free-living populations suggest that low folate status can also diminish genomic methylation, but primarily in individuals carrying the homozygous TT genotype of the common MTHFR 677C→T polymorphism. This nutrient–gene interaction highlights the importance of genetic background in determining susceptibility. Additional studies are needed to confirm whether folate-related hypomethylation in free-living populations is restricted to this genotype.
The tissue-specific sensitivity observed in these studies is biologically plausible. Bone marrow, with its extraordinarily high rate of DNA synthesis, appears particularly vulnerable to folate limitation. Similarly, the colonic mucosa, another tissue with rapid turnover, has been repeatedly identified as susceptible to folate depletion. This vulnerability may help explain why the epidemiological link between low folate intake and cancer risk is strongest for the colorectum.
Challenges in Detecting Folate Effects in Populations
Detecting folate’s impact on genomic methylation in the general population is more difficult than in controlled metabolic units. In ambulatory adults, three cross-sectional analyses found that folate status explained little of the variance in methylation. However, Friso and colleagues demonstrated a substantial effect when accounting for MTHFR genotype: individuals with the TT genotype and low folate status consistently exhibited diminished genomic methylation. This underscores the importance of considering nutrient–gene interactions in population studies.
Intervention Trials: Supplementation Outcomes
Several human intervention trials have examined whether folic acid supplementation can restore or enhance genomic methylation. Results are mixed:
- Cravo et al.:
- 10 mg/day folic acid significantly increased genomic methylation in normal rectal mucosa of subjects with prior adenomas or cancers.
- Later, 5 mg/day increased colonic methylation only in subjects with a single adenoma resected, not multiple.
- Kim et al.:
- 5 mg/day folic acid increased colonic methylation in subjects with resected adenomas.
- However, after 12 months, methylation levels in the placebo group rose to match supplementation—likely reflecting nationwide folic acid fortification during the trial.
- Physiological-range supplementation (400 μg/day):
- Increased methylation in blood leukocytes, but only a marginally significant effect (P = 0.09) in colonic mucosa.
- Higher-dose trials (700–2,000 μg/day):
- No effect on lymphocyte methylation.
- Notably, these null studies involved younger adults (mean ages 25 and 42), whereas positive trials included older populations (mean ages 56–66) with prior adenomas.
Age, Adenomas, and Sensitization to Folate
Age emerges as a strong inverse determinant of genomic methylation. Older subjects, particularly those with colonic adenomas, often exhibit regional hypomethylation surrounding neoplasms (a “field defect”). In such contexts, supplemental folic acid appears capable of increasing methylation, whereas in younger, healthy adults, supplementation has little effect.
This sensitizing effect of age and neoplastic lesions recapitulates findings in rodent models, where elder age predisposes tissues to folate-responsive methylation changes. Collectively, these observations suggest that folic acid supplementation may only enhance genomic methylation in human tissues already compromised by age-related decline or neoplastic perturbations.
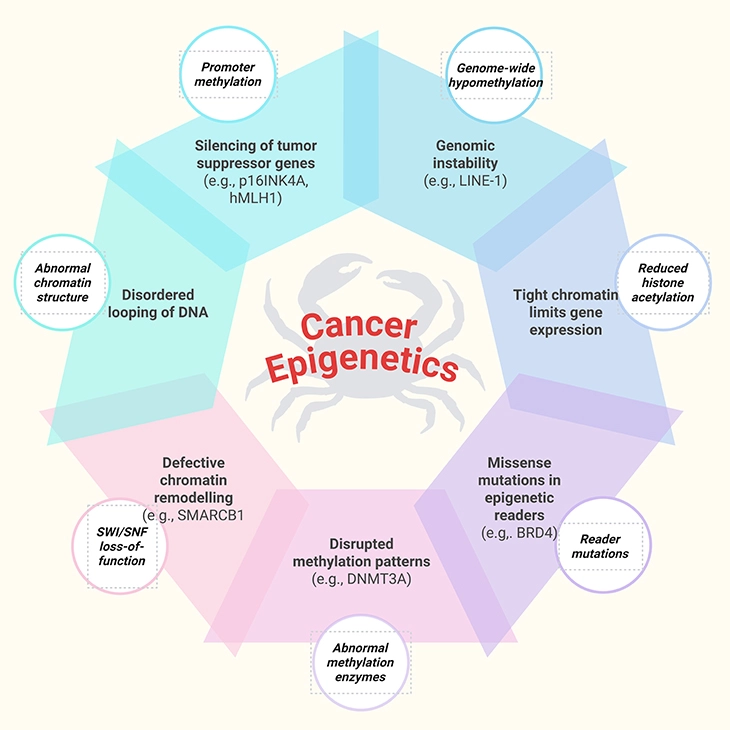
Figure 6. Cancer Epigenetics: How Altered Gene Regulation Fuels Tumor Development. How epigenetic changes can drive cancer, a simplified view of molecular disruption.
VI. Gene-Specific Methylation: Folate’s Dual Role in Carcinogenesis
Promoter Methylation and Tumor Suppressor Genes
Beyond global methylation, folate availability has been shown to influence gene-specific methylation, a phenomenon considered more relevant to the mechanistic evolution of carcinogenesis. In cell culture models, folate depletion induces hypermethylation of the promoter region of the putative tumor suppressor gene H-Cadherin, accompanied by a several-fold decrease in gene expression. Although counterintuitive, this repression aligns with observations that folate depletion increases steady-state mRNA for DNMT1 in cultured colonocytes, while levels of DNMT1, DNMT3a, and multiple methyl-CpG binding proteins rise in the livers of animals fed diets deficient in methionine, choline, and folate. These findings suggest that paradoxical site-specific hypermethylation may result from a compensatory up-regulation of the DNA methylation machinery in response to one-carbon nutrient depletion.
Clinical evidence remains limited but suggestive. In a cross-sectional study of head and neck cancers, low dietary folate intake was associated with a 2.3-fold increased risk of p16 promoter hypermethylation compared with tumors from individuals with higher folate intake. Similarly, promoter methylation of six “cancer-protective” genes—O6-MGMT, hMLH1, p14ARF, p16INK4A, RASSF1A, and Apc—was examined in colonic adenomas (see Figure 6). A consistent, though not statistically significant, trend toward less promoter hypermethylation was observed among those consuming >212 μg/day of folate.
In contrast, a placebo-controlled trial involving subjects with prior adenomas reported a different outcome. Participants receiving 5 mg folic acid plus 1.25 mg B12 daily for 6 months showed a trend (P = 0.08) toward greater combined promoter methylation across the six genes compared with controls. These mixed findings highlight the unresolved question of whether folate intake modulates promoter methylation in humans. Nevertheless, the emerging evidence supports the concept of a “dual effect” of folate—where both deficiency and excess may be deleterious, depending on context.
The Case of p53: Coding Region vs. Promoter
The p53 tumor suppressor gene has received particular attention, given its role in more than 85% of colorectal malignancies. Unlike many tumor suppressors, the p53 promoter lacks CpG islands, and thus hypermethylation of its promoter does not appear to suppress expression. Instead, diminished p53 function typically arises from loss of heterozygosity in one allele combined with a somatic mutation in the other.
Rodent studies confirm this distinction: severe folate deficiency reduced colonic p53 expression by ~40%, yet no changes were observed in methylation across 15 CpG sites in the promoter region.
In sharp contrast, hypomethylation of CpG sites in the coding region of p53 has been reproducibly induced by one-carbon nutrient depletion. Diets deficient in methionine, choline, and folate, isolated folate-deficient diets, and combined folate/B12/B6/B2-deficient diets all produced hypomethylation in the coding region. This effect has been observed in cultured human colonocytes and in the liver and colonic mucosa of rodents, particularly within the hypermutable region (exons 5–8)—the locus of most mutations in human cancers. The degree of hypomethylation correlates with the duration of folate depletion, though its causal role in carcinogenesis remains uncertain.
Paradoxically, it is the methylation—not demethylation—of these CpG sites that is thought to render them hotspots for C→T transition mutations, since spontaneous deamination of methylcytosine produces thymidine. Thus, folate’s impact on p53 methylation underscores the complexity of nutrient–gene interactions in cancer biology.
Other Mechanisms Beyond DNA Methylation
While nuclear DNA integrity and methylation remain the prevailing hypotheses linking one-carbon nutrients to carcinogenesis, alternative mechanisms may also contribute. Evidence is sparse, but possibilities include effects on cell-mediated immunity. However, such immune impairments have only been observed under severe, clinically irrelevant degrees of depletion, limiting their translational significance.
Final Reflection: Folate as a Double-Edged Sword
Taken together, the evidence suggests that folate and other one-carbon nutrients exert multifaceted effects on DNA methylation at both genomic and gene-specific levels. These effects are dynamic, tissue-specific, and context-dependent, shaped by factors such as age, genetic polymorphisms (e.g., MTHFR 677C→T), tissue turnover rates, and neoplastic processes.
The emerging picture is one of a “dual effect” of folate:
- Deficiency predisposes to genomic hypomethylation, gene-specific hypermethylation, and instability in critical loci such as p53.
- Excess supplementation, particularly in compromised tissues, may paradoxically enhance promoter hypermethylation of tumor suppressor genes.
Thus, folate functions as both a guardian and potential disruptor of genomic integrity, underscoring the delicate balance required for optimal one-carbon metabolism in cancer prevention (see Figure 6).
Take-Home Messages
- Folate is a genomic gatekeeper: Adequate folate sustains the SAM-dependent methylation cycle, preserving DNA integrity and regulating gene expression.
- Balance matters more than extremes: Both deficiency and excess supplementation can disrupt methylation—manifesting as global hypomethylation or gene-specific hypermethylation.
- Genomic vs. gene-specific effects diverge: While global hypomethylation destabilizes chromosomal architecture, promoter hypermethylation silences tumor suppressor genes, each contributing uniquely to carcinogenesis.
- Genetic background modifies risk: The MTHFR 677C→T polymorphism (TT genotype) amplifies the impact of low folate status, highlighting the importance of nutrient–gene interactions.
- Tissue turnover dictates vulnerability: Rapidly proliferating tissues such as bone marrow and colonic mucosa are especially sensitive to folate depletion, explaining the strong link between folate status and colorectal cancer risk.
- Age is a sensitizer: Elder age predisposes tissues to hypomethylation, making them more responsive to folate modulation—whether protective or deleterious.
- p53 illustrates complexity: Folate depletion induces hypomethylation in coding regions of p53, but not its promoter, underscoring the gene-specific nuances of methylation in cancer evolution.
- Dual effect paradigm: Folate acts as both protector and potential disruptor—its role in carcinogenesis depends on dose, duration, genetic context, and tissue environment.
Summary and Conclusions
Folate-dependent one-carbon metabolism is a cornerstone of cellular physiology, linking nutrient availability to DNA synthesis, repair, and methylation. Our synthesis highlights the dual effect paradigm: folate deficiency predisposes to genomic hypomethylation, gene-specific hypermethylation, and instability in critical loci such as p53, while excess supplementation may paradoxically enhance promoter hypermethylation of tumor suppressor genes. Evidence from cell culture, animal models, and human intervention trials underscores the complexity of folate’s role in carcinogenesis, shaped by dose, duration, genetic polymorphisms (e.g., MTHFR 677C→T), tissue turnover rates, and age-related susceptibility. The strongest epidemiological links emerge in rapidly proliferating tissues such as the colonic mucosa, explaining why diminished folate intake most consistently associates with colorectal cancer risk.
Despite decades of research, critical gaps remain. First, the precise mechanisms by which folate depletion simultaneously induces global hypomethylation and site-specific hypermethylation are unresolved. Second, the thresholds of folate intake that distinguish protective from deleterious effects remain poorly defined, particularly in vulnerable populations such as children with autism spectrum disorder receiving folinic acid supplementation. Third, while SAM/SAH ratios are widely used as proxies for methylation potential, their predictive value is inconsistent, reflecting the influence of additional regulatory factors.
Recent literature expands these concerns. Advances in DNA methylation profiling technologies highlight the potential of methylation hotspots as biomarkers for cancer diagnosis and prognosis, yet translation into routine clinical practice remains limited. In colorectal cancer, methylation of key genes such as hMLH1 and O6-MGMT is increasingly recognized as both a driver of tumorigenesis and a potential therapeutic target, with DNA methylation inhibitors emerging as novel strategies. Moreover, folate metabolism is now implicated not only in cancer but also in neurodegenerative diseases, suggesting broader relevance across human health.
Future Directions
Future research must address several priorities:
- Defining safe supplementation thresholds: Longitudinal studies are needed to clarify whether supraphysiological folate or folinic acid intake—especially in genetically predisposed or pediatric populations—modulates cancer risk.
- Integrating epigenetic biomarkers into clinical practice: Large-scale trials should validate DNA methylation signatures as tools for early detection, prognosis, and personalized therapy.
- Exploring tissue-specific responses: Comparative studies across tissues with varying turnover rates will help explain why some organs (e.g., colon, bone marrow) are more vulnerable to folate perturbations.
- Expanding beyond cancer: Investigations into folate’s role in neurodevelopment and neurodegeneration may reveal overlapping pathways that connect metabolism, epigenetics, and disease.
In conclusion, folate remains both a guardian and potential disruptor of genomic integrity. Precision in supplementation—guided by genetic background, age, tissue context, and emerging biomarkers—is essential to harness its protective effects while minimizing unintended risks.
“Folate is both a guardian and a gamble—its power to protect or to disrupt rests in the balance of dose, context, and time.”
Did You Know? Folate Receptor Autoantibodies (FRAAs) may impede proper folate transport.
Folate (vitamin B9) is very important for your child’s brain development!
During pregnancy, it helps prevent neural tube defects and plays a big role in forming a normal and healthy baby’s brain and spinal cord. Folate also helps cells divide and assists in both DNA and RNA synthesis.
Emerging research suggests that the presence of FRAAs negatively impacts folate transport into the brain.
- Recent studies reveal that a large subgroup of children with autism spectrum disorder (ASD) have FRAAs.
- This suggests that a possible disruption in folate transport across the blood-cerebrospinal fluid (CSF) barrier may potentially influence ASD-linked brain development.
- Screening for the FRAAs in your child should be part of your early intervention strategies.
Is there a test for identifying Folate Receptor Autoantibodies (FRAAs)?
Yes, there is a test – The Folate Receptor Antibody Test (FRAT®) has emerged as a diagnostic tool for detecting the presence of FRAAs.
It is important to screen at an early age or as soon as possible as there may be corrective measures available. Please consult your physician for further information.
To request a test kit, click on the button below.

For information on autism monitoring, screening and testing please read our blog.
References
- de la Torre Guzmán SR, Pelayo-Chávez B, García-Muro AM, Soto-Reyes E, Sánchez-López JY. The Role of Folic Acid in DNA Methylation and Breast Cancer. Int J Vitam Nutr Res. 2025 Mar 28;95(2):26221. doi: 10.31083/IJVNR26221. PMID: 40298153.
https://pubmed.ncbi.nlm.nih.gov/40298153/
→ This review explores folate metabolism, DNA methylation, and breast cancer risk, highlighting both protective and deleterious effects. - Prinz-Langenohl R, Fohr I, Pietrzik K. Beneficial role for folate in the prevention of colorectal and breast cancer. Eur J Nutr. 2001 Jun;40(3):98-105. doi: 10.1007/pl00007387. PMID: 11697447.
https://pubmed.ncbi.nlm.nih.gov/11697447/
→This review summarizes the epidemiological evidence for the association between folate status and colorectal and breast cancer risk. - Kim YI. Folate and colorectal cancer: an evidence-based critical review. Mol Nutr Food Res. 2007 Mar;51(3):267-92. doi: 10.1002/mnfr.200600191. PMID: 17295418.
https://pubmed.ncbi.nlm.nih.gov/17295418/
→ An evidence-based critical examination the role of folate in the development and progress of colorectal cancer (CRC). - Kim YI. Folate and DNA methylation: a mechanistic link between folate deficiency and colorectal cancer? Cancer Epidemiol Biomarkers Prev. 2004 Apr;13(4):511-9. PMID: 15066913.
https://pubmed.ncbi.nlm.nih.gov/15066913/
→ Classic mechanistic review detailing how folate deficiency alters methylation patterns, contributing to carcinogenesis. - Kim YI. Nutritional epigenetics: impact of folate deficiency on DNA methylation and colon cancer susceptibility. J Nutr. 2005 Nov;135(11):2703-9. doi: 10.1093/jn/135.11.2703. PMID: 16251634.
https://pubmed.ncbi.nlm.nih.gov/16251634/
→ Suggesting that the effects of folate deficiency and supplementation on DNA methylation are gene and site specific, and appear to depend on cell type, target organ, stage of transformation, and the degree and duration of folate depletion. - Ye M, Xu G, Zhang L, Kong Z, Qiu Z. Meta Analysis of Methylenetetrahydrofolate Reductase (MTHFR) C677T polymorphism and its association with folate and colorectal cancer. BMC Cancer. 2025 Jan 29;25(1):169. doi: 10.1186/s12885-025-13546-w. PMID: 39875876; PMCID: PMC11776141.
https://pubmed.ncbi.nlm.nih.gov/39875876/
→ Systematic review of 100 studies (39,702 cases, 55,718 controls) linking MTHFR polymorphisms, folate intake, and colorectal cancer risk. - Crider KS, Yang TP, Berry RJ, Bailey LB. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate’s role. Adv Nutr. 2012 Jan;3(1):21-38. doi: 10.3945/an.111.000992. Epub 2012 Jan 5. PMID: 22332098; PMCID: PMC3262611.
https://pubmed.ncbi.nlm.nih.gov/22332098/
→ Broad review connecting folate metabolism, epigenetic regulation, and disease outcomes, including cancer and neurodevelopment. - Stover PJ, James WPT, Krook A, Garza C. Emerging concepts on the role of epigenetics in the relationships between nutrition and health. J Intern Med. 2018 Jul;284(1):37-49. doi: 10.1111/joim.12768. Epub 2018 May 23. PMID: 29706028.
https://pubmed.ncbi.nlm.nih.gov/29706028/
→ New emerging concepts at the interface of nutrition and epigenetics were discussed. - Friso S, Choi SW. Gene-nutrient interactions in one-carbon metabolism. Curr Drug Metab. 2005 Feb;6(1):37-46. doi: 10.2174/1389200052997339. PMID: 15720206.
https://pubmed.ncbi.nlm.nih.gov/15720206/
https://www.benthamdirect.com/content/journals/cdm/10.2174/1389200052997339
→ Review highlighting nutrient–gene interactions (MTHFR, DNMTs) and their impact on methylation and cancer risk. - Hoxha B, Hoxha M, Domi E, Gervasoni J, Persichilli S, Malaj V, Zappacosta B. Folic Acid and Autism: A Systematic Review of the Current State of Knowledge. Cells. 2021 Aug 3;10(8):1976. doi: 10.3390/cells10081976. PMID: 34440744; PMCID: PMC8394938.
https://pubmed.ncbi.nlm.nih.gov/34440744/
→ Systematic review not only elucidating the potential role of folic acid in autism spectrum disorders but also investigating various mechanisms that are involved.



Comments are closed.