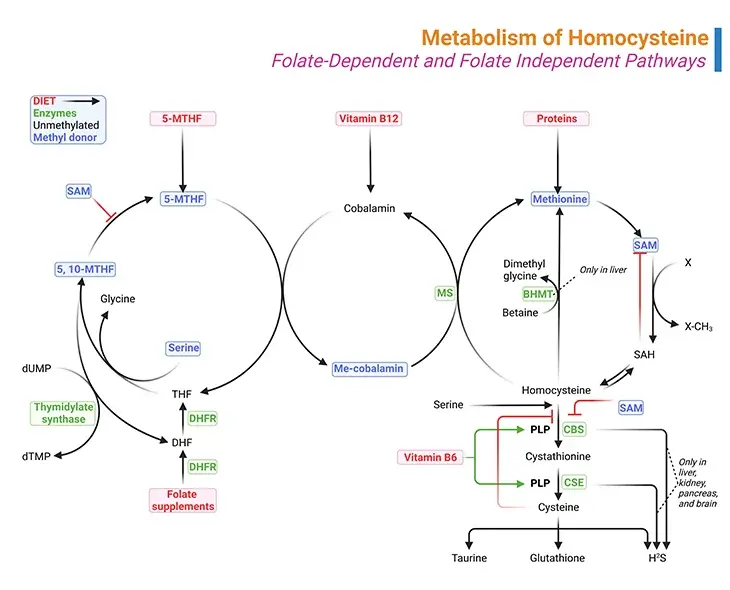
Figure 1. Metabolism of Homocysteine. Metabolism of homocysteine involves folate-dependent and folate-independent pathways. (i) A deficiency of the enzyme cystathionine beta-synthase (CBS), methylenetetrahydrofolate reductase (MTHFR), and methionine synthase (MS) results in the accumulation of homocysteine. (ii) Similarly, vitamin deficiencies in folate (vitamin B9), cobalamin (vitamin B12), or pyridoxine (vitamin B6) can also result in the accumulation of homocysteine. (iii) Methylmalonyl-CoA mutase also requires vitamin B12 as a cofactor and therefore a deficiency in vitamin B12 will result in the accumulation of methylmalonic acid as well.
Introduction
1. Developmental Delays: The birth of a child is a marvelous event, and the parents wonder about the birth of this tiny person; and have hopes and dreams for its future as it journeys through life. Nevertheless, it is equally hard to understand the tremendous complexity of this life form, as it blossoms. Since the embryological development is so complex and most of the time, the intricacies of biochemical and neurological development of the child necessitate that errors do occur.
Etiological factors in developmental delays can be genetic, developmental, environmental, or a combination [1]. For instance:
- Genetic causes include chromosomal and inherited conditions (e.g., errors in genetic code or epigenetic code)
- Developmental and environmental factors include prenatal exposure to infections and toxins
- Environmental or acquired factors include prenatal trauma (e.g., prematurity)
- Sociocultural actors
- Deprivation of nutrition, nurturance and social stimulation.
Current knowledge suggests that genetic, environmental, biologic, and psychosocial factors work additively to the emergence of delay in normal development and quite often lifelong disability. A child may have a developmental delay when a child’s overall development progresses more slowly than normal, and it eventually persists in one or more areas (neurodevelopmental domains), such as: (i) learning, (ii) language, (iii) behavior, and (iv) motor skills.
These conditions appear during the developmental period and ultimately lead to disabilities that may impair a person not only physically but also intellectually throughout the individual’s lifetime [1] (see Figure 2).
2. Intellectual disability (ID): ID formerly known as mental retardation, can be caused by a range of environmental and genetic factors that lead to a combination of cognitive and social impairments. In other words, ID is a neurodevelopmental disorder that is characterized by significant limitations of both intellectual function and in adaptive behavior [2, 7] (see Figure 2).
The prevalence of ID at any one time is estimated to range from 1 to 3 percent of the population in Western societies with a major impact on the affected individuals, loved ones, as well as the health care system. The highest incidence of ID is reported in school-aged children, with a peak at ages 10 to 14 years old. ID is about 1.5 times more common among males than females.
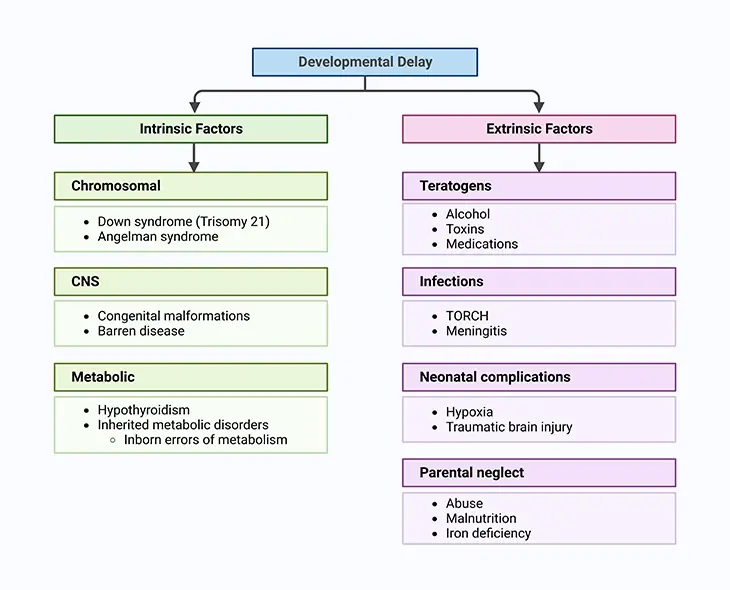
Figure 2. Factors Leading to both Physical and Intellectual Development in the Child. Current knowledge suggests that genetic, environmental, biologic, and psychosocial factors work additively to the emergence of delay in normal development and quite often lifelong disability.
In children, ID is the third most frequent neurological conditions after seizure (epilepsy) and cerebral palsy (motor disability). Seizure disorders occur more commonly in individuals with IDs than in the general population. An evaluation of psychiatric disorders in children and adolescents with ID and epilepsy found that approximately one-third had co-morbid autism spectrum disorders (ASDs). The combination of intellectual disability, epilepsy, and ASD may occur in 0.07% of the general population [6, 7]. Consequently, establishing etiologies for ID is a major challenge.
3. Inborn errors of metabolism (IEMs): IEMs accounts for a small percentage (<0.5%) of children with unexplained ID. IEMs are rare genetic or inherited disorders resulting from an enzyme defect in biochemical and metabolic pathways affecting proteins, fats, and carbohydrates metabolism or impaired organelle (lysosome, peroxisome, and mitochondria) function manifesting as complicated medical conditions involving several human organ systems. Thus, IEMs are disorders in which a single gene defect results in accumulation of enzyme substrates behind a metabolic obstruction or deficiency of a reaction product. The clinical presentation of IEMs are quite complex and differ widely with both mild and severe forms depending on the remaining activity of the altered enzyme [3] (see Figure 2).
Age of presentation can vary from infancy to adolescence with the more severe forms appearing in early childhood accompanied by significant morbidity and mortality. While most IEMs occur early in life, generally, the metabolism of the mother may compensate for many IEMs while in utero and so the symptoms first appear in the neonatal period.
IEMs exemplify the largest category of single gene disorders amenable to causal therapy. Furthermore, early detection of IEMs is crucial for:
- improving outcomes,
- preventing adverse manifestations, and
- reducing burden on affected individuals and their families.
More importantly, (a) as IEMs are being diagnosed at earlier ages resulting in the extension of lives that may have been lost, (b) the number of women with IEMs who are now reaching child-bearing age is increasing, which may adversely affect the fetus.
Even though, taken individually, each IEM disorder is undoubtedly quite rare indeed. However, considering the sheer number of possible mutations that have been defined thus far, in the aggregate, IEMs represents a substantial number of potential cases, approximately 1:1500 children, and a daunting challenge to clinicians to comprehend and diagnose.
Metabolism of Homocysteine (Hcy)
The biologically important amino acid homocysteine links sulfur, methionine, and one-carbon metabolism. Homocysteine is a non-essential, sulfur containing, non-proteinogenic amino acid. It is synthesized by transmethylation of the essential, diet-derived amino acid methionine. It represents the only way through which homocysteine is produced in humans [4].
Since homocysteine identification 90 years ago, several research reports have described its relevance to several IEMs, B vitamin status, and disease as diverse as cardio- and cerebrovascular disease, dementia, renal disease, thyroid disease, and pregnancy complications, to name but a few.
1. Role of methionine metabolism in the metabolic cycles of folate and homocysteine
Methionine is an essential amino acid found in meat, fish, and dairy products. Amino acids are the building blocks used to make proteins. Methionine cannot be synthesized by the body; therefore it must be consumed in the diet.
Methionine, like other amino acids, has a dual fate:
- One that involves protein synthesis, and
- The other involves its catabolism giving rise to many sulfur-containing compounds, for example, S-adenosylmethionine (SAM), S-adenosyl-L-homocysteine (SAH), homocysteine (Hcy), cystathionine, and cysteine (see Figure1).
The (i) folate cycle, (ii) methionine cycle, and (iii) transsulfuration pathway are three interdependent pathways, essential for the synthesis of these molecules (see Figure 1).
(i) Methionine Cycle
The conversion of methionine to cysteine begins with combining methionine with ATP to form SAM, which is one of the primary methyl donors for methylation reactions in the body, including methylation of lipids, DNA and neurotransmitters.
More than 35 reactions in the human body require methyl donation from SAM. With the transfer of its methyl group, SAM is converted to SAH as shown in Figure 1. The next step is the hydrolysis of SAH to homocysteine and adenosine. At this point, homocysteine may be remethylated back to methionine or condensed in transsulfuration with serine to form cystathionine.
(ii) Folate Cycle
Remethylation of homocysteine is primarily carried out by methionine synthase (MS), which requires methyl-cobalamin (Me-cobalamin) as a cofactor. Cobalamin is methylated by 5-methyltetrahydrofolate (5-MTHF) forming methyl-cobalamin and tetrahydrofolate (THF). The recycling of 5-MTHF involves the folate cycle [5].
Tetrahydrofolate (THF) is converted to 5, 10-methylenetetrahydrofolate (5, 10-Methylene-THF) by methylenetetrahydrofolate reductase (MTHFR) and then to 5-MTHF. Note that MTHFR is a flavin adenine dinucleotide-dependent enzyme (FAD).
The liver contains a second pathway to remethylate homocysteine to methionine that uses betaine as the methyl donor and the enzyme betaine methyltransferase (BHMT), as illustrated in Figure 1.
(iii) Transsulfuration Pathway
The transsulfuration pathway occurs primarily in the liver and kidney. Condensation of homocysteine with serine is catalyzed by cystathionine beta-synthase (CBS), a pyridoxal phosphate (PLP, vitamin B6)-dependent enzyme, to form cystathionine in a transsulfuration reaction.
Cystathionine is then broken down to cysteine though another transsulfuration reaction by another pyridoxal phosphate-dependent enzyme, viz., gamma-cystathionase (CSE).
Therefore, the folate cycle, methionine cycle, and transsulfuration pathway depend on several B-vitamins, viz., folate (B9), riboflavin (B2), cobalamin (B12), and pyridoxal phosphate (B6).
Clinical Significance
It is important to note that cysteine is a conditionally essential amino acid as it is made from the essential amino acid methionine (see Figure 2). Therefore, folate cycle, methionine cycle, and transsulfuration pathways are interdependent on one another, defects in any one of these pathways may affect cysteine levels. Apart from being an amino acid for protein synthesis, cysteine is an important precursor of the antioxidant glutathione. Glutathione is important as a reductant, detoxifying agent, and in the transport of amino acids.
2. Disorders of the metabolic cycles of folate, methionine, and homocysteine
Under normal conditions, homocysteine levels are very low in the serum as it is either remethylated to methionine or catabolized to cysteine in several steps. The enzymes involved in these reactions require adequate stores of (a) vitamin B9 (folic acid), (b) vitamin B12 (cobalamin), and (c) vitamin B6 (pyridoxal phosphate). Inadequate levels of these vitamins or any defect in enzymes along these metabolic pathways may result in homocystinuria.
However, the most common cause of homocystinuria is a defect in the pyridoxal phosphate-dependent CBS enzyme, which transforms homocysteine to cystathionine. In the absence of this enzyme, homocysteine and methionine concentrations dramatically increase in the serum and spill into the urine, leading to homocystinuria.
More importantly, homocysteine is toxic to neural tissue in high concentrations. Thus, a child with this inborn error has normal brain function at birth but experiences slowly progressive neurological injury should the metabolic derangement go unrecognized.
Increased homocysteine is also associated with accelerated vascular disease and increased risk of stroke, for example, cerebral venous thrombosis [6]. About 50% of these patients respond to pyridoxal phosphate therapy, which can increase the activity of the defective enzyme.
Elevated plasma and urine homocysteine levels indicate classical homocystinuria due to CBS deficiency. However, CBS deficiency is a fairly rare IEM and there are many other genetic, nutritional, and pharmacological factors, as well as several diseases associated with increased homocysteine.
Vitamin B12 and, less often, folate deficiency are common nutritional causes of elevated homocysteine concentrations. A vitamin B12 deficiency can be excluded if methylmalonic acid is not detected in the urine.
Due to folate fortification of food since 1998, folate deficiency is rare in the United States. However, folate deficiency may develop with elevated levels of homocysteine due to homozygous variant, [c.677C>T] variant of the MTHFR gene. Unlike CBS deficiency, homocystinuria may occur without elevation of plasma methionine as the product of the MTHFR reaction, 5-MTHG, is required to form methyl cobalamin. In addition, genetic testing is needed to confirm the deficiency of cystathionine beta-synthase.
There are also pharmacological factors, such as methotrexate treatment, which may reduce folate levels.
Take home messages
- Hyperhomocysteinemia indicates high homocysteine levels in the blood. It should not be confused with homocystinuria.
- Cystathionine beta-synthase (CBS) deficiency is the only defect that consistently causes homocystinuria.
- Homocysteine can either be remethylated to methionine or undergo transsulfuration to cysteine. This second step is regulated by cystathionine beta-synthase and utilizes vitamin B6 (pyridoxine) as an essential co-factor.
- Cystathionine beta-synthase deficiency is a rare autosomal recessive disorder. It can result in early developmental delays. Later, it may present with phenotypic abnormalities resembling Marfan syndrome, osteoporosis, ectopia lentis, and recurrent thrombosis. Developmental delays in the newborn or child distinguish this entity from Marfan syndrome.
- Other causes of homocysteinemia include deficiencies in vitamin B12, vitamin B6, and folate, inborn errors of homocysteine remethylation, renal insufficiency, and medications.
Conclusion
In this blog we have described succinctly the basics of homocysteine metabolism and the factors that influence it. This general overview of homocysteine: considers the diseases, disorders, and conditions associated with impaired homocysteine metabolism, and its relevance pertaining to several IEMs, B vitamin status, and a plethora of diseases as diverse as cardio- and cerebrovascular diseases. As well as homocysteine metabolism and its link to homocysteinemia- and homocystinuria-related pathologies in humans.
Did You Know? Folate Receptor Autoantibodies (FRAAs) may impede proper folate transport.
Folate (vitamin B9) is very important for your child’s brain development!
During pregnancy, it helps prevent neural tube defects and plays a big role in forming a normal and healthy baby’s brain and spinal cord. Folate also helps cells divide and assists in both DNA and RNA synthesis.
Emerging research suggests that the presence of FRAAs negatively impacts folate transport into the brain.
- Recent studies reveal that a large subgroup of children with autism spectrum disorder (ASD) have FRAAs.
- This suggests that a possible disruption in folate transport across the blood-cerebrospinal fluid (CSF) barrier may potentially influence ASD-linked brain development.
- Screening for the FRAAs in your child should be part of your early intervention strategies.
Is there a test for identifying Folate Receptor Autoantibodies (FRAAs)?
Yes, there is a test – The Folate Receptor Antibody Test (FRAT®) has emerged as a diagnostic tool for detecting the presence of FRAAs.
It is important to screen at an early age or as soon as possible as there may be corrective measures available. Please consult your physician for further information.
To request a test kit, click on the button below.

For information on autism monitoring, screening and testing please read our blog.
References
- Choo YY, Agarwal P, How CH, Yeleswarapu SP. Developmental delay: identification and management at primary care level. Singapore Med J. 2019 Mar;60(3):119-123. doi: 10.11622/smedj.2019025. PMID: 30997518; PMCID: PMC6441684.
https://pubmed.ncbi.nlm.nih.gov/30997518/ - Schalock RL, Luckasson R, Tassé MJ. An Overview of Intellectual Disability: Definition, Diagnosis, Classification, and Systems of Supports (12th ed.). Am J Intellect Dev Disabil. 2021 Nov 1;126(6):439-442. doi: 10.1352/1944-7558-126.6.439. PMID: 34700345.
https://pubmed.ncbi.nlm.nih.gov/?term=Schalock+RL&cauthor_id=34700345/ - Agana M, Frueh J, Kamboj M, Patel DR, Kanungo S. Common metabolic disorder (inborn errors of metabolism) concerns in primary care practice. Ann Transl Med. 2018 Dec;6(24):469. doi: 10.21037/atm.2018.12.34. PMID: 30740400; PMCID: PMC6331353.
https://pubmed.ncbi.nlm.nih.gov/30740400/ - Škovierová H, Vidomanová E, Mahmood S, Sopková J, Drgová A, Červeňová T, Halašová E, Lehotský J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int J Mol Sci. 2016 Oct 20;17(10):1733. doi: 10.3390/ijms17101733. PMID: 27775595; PMCID: PMC5085763.
https://pubmed.ncbi.nlm.nih.gov/27775595/ - Hoxha B, Hoxha M, Domi E, Gervasoni J, Persichilli S, Malaj V, Zappacosta B. Folic Acid and Autism: A Systematic Review of the Current State of Knowledge. Cells. 2021 Aug 3;10(8):1976. doi: 10.3390/cells10081976. PMID: 34440744; PMCID: PMC8394938.
https://pubmed.ncbi.nlm.nih.gov/34440744/ - Petras M, Tatarkova Z, Kovalska M, Mokra D, Dobrota D, Lehotsky J, Drgova A. Hyperhomocysteinemia as a risk factor for the neuronal system disorders. J Physiol Pharmacol. 2014 Feb;65(1):15-23. PMID: 24622826.
https://pubmed.ncbi.nlm.nih.gov/24622826/ - Smith AD, Refsum H. Homocysteine, B Vitamins, and Cognitive Impairment. Annu Rev Nutr. 2016 Jul 17;36:211-39. doi: 10.1146/annurev-nutr-071715-050947. PMID: 27431367.
https://pubmed.ncbi.nlm.nih.gov/27431367/


