Table of Contents
- Introduction
- The Biopterin System, Coenzyme of Aromatic Amino Acid Hydroxylases
- Epidemiology
- Clinical Features of HPA
- Diagnosing HPA
- Treating HPA
- The Relationship Between Biopterin and Folate Metabolism
- Tetrahydrobiopterin (BH4) Deficiency in ASD
- Take Home Messages
- Summary and Conclusions
- Did You Know About Folate Receptor Autoantibodies (FRAAs) and Brain Development?
- References
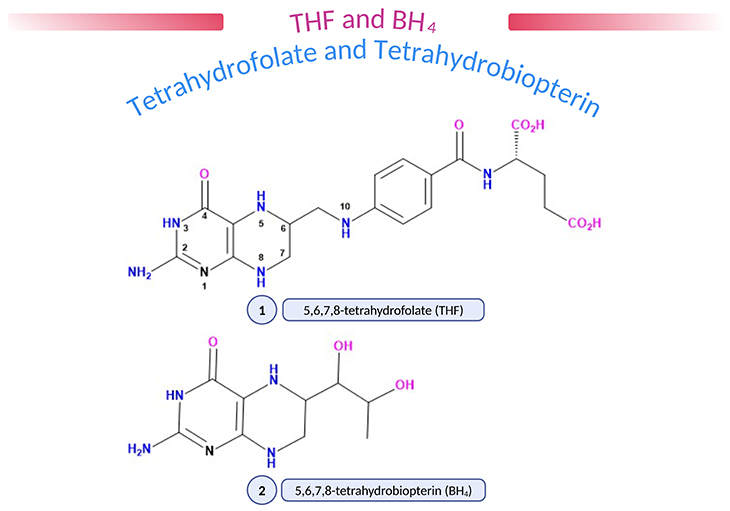
Figure 1. (1) 5,6,7,8-tetrahydrofolate (THF); (2) 5,6,7,8-tetrahydrobiopterin (BH4). Natural pteridines contain a 2-amino,4-oxopteridine (or ‘pterin’) ring. Folic acid and biopterin contain a fully oxidized pterin ring (five double bonds) but in vivo the number and disposition of double bonds and protons are in the dihydro- or tetrahydro- form (as shown above), enabling pteridines to act as proton donors and acceptors. Various substituent groups may be attached at position 6, biopterins having a dihydroxypropyl, and folate a methylene/p-aminobenzoate/glutamate group. Folates may carry additional methyl (CH3), formyl (CHO), or formimino (CHNH) groups at N5, CHO at N10 or methenyl (CH) or methylene (CH2) groups linking N5 and N10, reflecting the role of folate in 1-carbon (1-C) transfer. Amethopterin (methotrexate) is a synthetic pteridine which differs from folic acid in having an amino-group instead of an oxo-group at position 4 and a methyl group at position 10.
Introduction
Phenylketonuria (PKU) is a rare but potentially serious inherited disorder. Our bodies break down the protein in foods, such as meat and fish, into amino acids, which are the ‘building blocks’ of protein. These amino acids are then utilized to make our own new proteins. Any amino acids that are not required are broken down further and excreted from the body. Individuals with PKU cannot break down the amino acid phenylalanine (PA), which in turn builds up in their blood and brain. This can lead to brain damage.
The Biopterin System, Coenzyme of Aromatic Amino Acid Hydroxylases
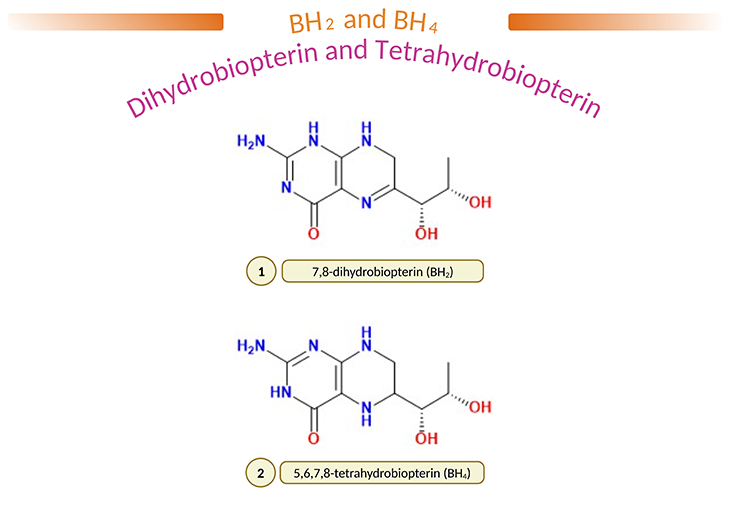
Figure 2. (1) 7,8-dihydrobiopterin (BH2) and (2) 5,6,7,8-tetrahydobiopterin (BH4). L-erythro-5,6,7,8-tetrahydrobiopterin (BH4) is a common natural cofactor for hydroxylases of phenylalanine, tyrosine, and tryptophan, and is thought to play an important role in the regulation of monoamine neurotransmitter biosynthesis. In fact, it is well known that in ‘atypical phenylketonuria,’ BH4 deficiency results in severe neurological problems as result of decreased biosynthesis of brain catecholamines and 5-hydroxytrptamine (5-HT). Furthermore, it has been reported that the content of BH4 is reduced in the brain and/or cerebrospinal fluid (CSF) of several neuropsychiatric diseases, such as Parkinson’s disease, Alzheimer’s disease, depression, and dystonia; and most importantly, in autism spectrum disorders (ASD). Administration of BH4 improve clinical signs and symptoms of these diseases; and therapeutic efficacy are mainly due to enhancement of neurotransmitter release.
PKU is a metabolic disorder that is inherited in an autosomal recessive fashion and is caused by a defect in the hepatic PA hydroxylating system [1]. This system consists of two enzymes, viz.:
- phenylalanine 4-hydroxylase (PAH), and
- dihydropteridine reductase (DHPR),
as well as a coenzyme, tetrahydrobiopterin (BH4) (see Figure 1 and 2), which acts not only on PAH but also on, for instance:
- tyrosine 3-hydroxylase (TH), and
- tryptophan 5-hydroxylase (TPH).
The former on the ‘dopamine and norepinephrine synthesis pathway;’ and the later on the ‘serotonin synthesis pathway’ (see Figure 3). (cf. previous blogs entitled as: (i) ‘The Neurotypical Brain versus the Autistic Brain: Neurotypical Brain Chemistry,’ (ii) ‘The Neurotypical Brain versus the Autistic Brain: Brain Chemistry in ASD.’).
During hydroxylation cycle of these three enzymes (PAH, TH, TPH), BH4 is oxidized to quinonoid dihydrobiopterin (q-BH2), the latter being transformed partly into L-erthro-7,8-dihydrobiopterin (7,8-BH2). They are further reduced to BH4 by dihydropteridine reductase (DHPR) and by dihydrofolate reductase (DHFR), respectively (see Figure 3) [2].
The classic form of PKU is caused by a lack of PAH activity, which normally transforms PA into tyrosine. This defect produces hyperphenylalaninemia (HPA) which, if left untreated, results in severe mental disability (cf. previous blog entitled as: ‘Developmental Delays in Infancy and Early Childhood: Phenylalanine and Tyrosine Metabolism Disorders.’).
Moreover, hyperphenylalaninemia impedes the transfer of L-tryptophan and L-tyrosine at the blood-brain barrier (BBB) level. This results in a decreased synthesis of ‘biogenic amines.’ Additionally, increased levels of PA in brain seems to directly inhibit TH and TPH activities. The correction of HPA with a low-PA diet leads to a normal biogenic amine synthesis. Neonatal screening for HPA and early low-PA diet allow a normal development in children with PKU.
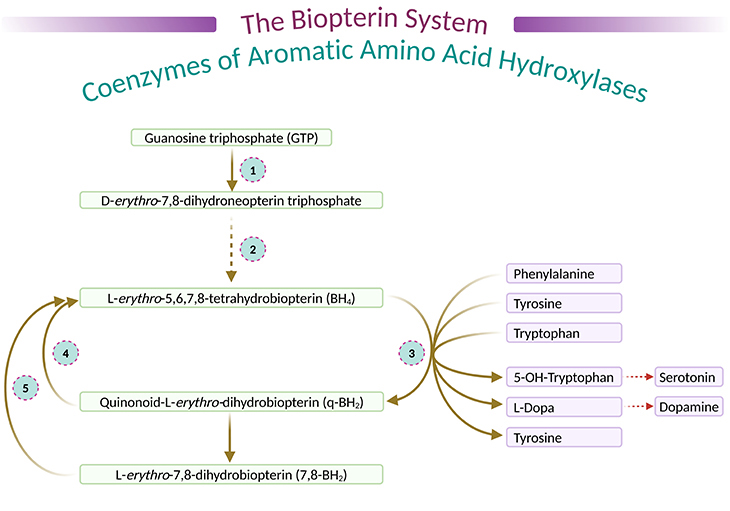
Figure 3. The biopterin system, coenzymes of aromatic amino acid hydroxylase. (1) Guanosine triphosphate cyclohydrolase; (2) dihydrobiopterin synthetase; (3) phenylalanine 4-hydroyxlase, tyrosine 3-hydroxylase, tryptophan 5-hydroxylase; (4) dihydropteridine reductase; and (5) dihydrofolate reductase.
[L-erythro-7,8-dihydrobiopterin – a 7,8-dihydrobiopterin in which the 1,2-dihydroxypropyl group has (1R,2S)-configuration, and a naturally occurring form – It is an enantiomer of a D-erythro-7,8-dihydrobiopterin; D-erythro-7,8-dihydrobiopterin in which the 1,2-dihydroxypropyl group has (1S,2R)-configuration; 6R-L-erythro-5,6,7,8-tetrahydrobiopterin – 5,6,7,8-tetrahydrobiopterin in which the 1,2-dihydroxypropyl side-chain has L-erythro-configuration.]
Epidemiology
PKU is one of the most frequent inherited disorders in Europeans; and the reported incidence of PKU from newborn screening programs ranges from 1 in 13,500 to 19,000 newborns in the United States. Some 1.5% – 2% of PKU cases are not due to a defect in PAH but are due to a defect in BH4 synthesis or in the regeneration of BH4 (DHPR deficiency; see Figure 4).
Clinical Features of HPA
In either case, the result is the development of ‘malignant HPA,’ that is PKU which does not respond to a good dietary control of blood-PA levels and leads to an irreversible neurologic deterioration that becomes apparent between 3 to 5 months of age. This is characterized by the following clinical features, for example:
- myoclonic seizures, axial hypotonia, extrapyramidal hypertonia with lead-pipe rigidity of upper limbs, and distal tremor;
- oculo motor dysfunction with episodes of upper and sideward gaze deviation;
- expressionless facies, buccofacial dyskinesia and drooling, and swallowing difficulties.
This deterioration which occurs in spite of satisfactory blood-PA levels is due to biogenic amine deficiency, viz., serotonin, dopamine, and norepinephrine. Death typically occurs by age 5 – 7 years.
Diagnosing HPA
Of paramount importance is early recognition of these cases. ‘DHPR and biopterin assay ’ on dried blood spots and biopterin assay on filter paper spots of urine should be carried out in every infant with a positive neonatal screening for HPA.
Treating HPA
Positive results require dietary control of blood-PA levels, a supplementation with neurotransmitter precursor, such as L-dopa and 5-hydroxy tryptophan (5-HT) (with a peripheral inhibitor of aromatic amino acid decarboxylase, carbidopa). As well as in cases of BH4 synthesis deficiency, a replacement therapy with BH4.
Nevertheless, it has recently become evident that the treatment of DHPR deficiency is not fully satisfactory if folinic acid (5-formyl-tetrahydorfolate, [THF]) supplementation is not started early. DHPR-deficient patients has led us to a better understanding of ‘the relationship between folate and biopterin metabolism.’ In such patients, folic acid deficiency in blood and brain seems usual, with development of basal ganglia calcifications, as in cases of inherited defect of folate transfer into brain [3].
In addition, some cases of DHPR deficiency with good dietary control of blood-PA levels, sufficient neurotransmitters precursor supplementation, and normal neurologic status; have developed at age 2 – 3 years, an unexpected neurologic deterioration with signs and symptoms similar to early manifestations of untreated cases and associated with decreased levels of CSF amine metabolites. This late neurologic deterioration is secondary to brain and CSF 5-methyl-THF deficiency; and has been cured by 5-formyl-THF (folinic acid) in occasional patients.
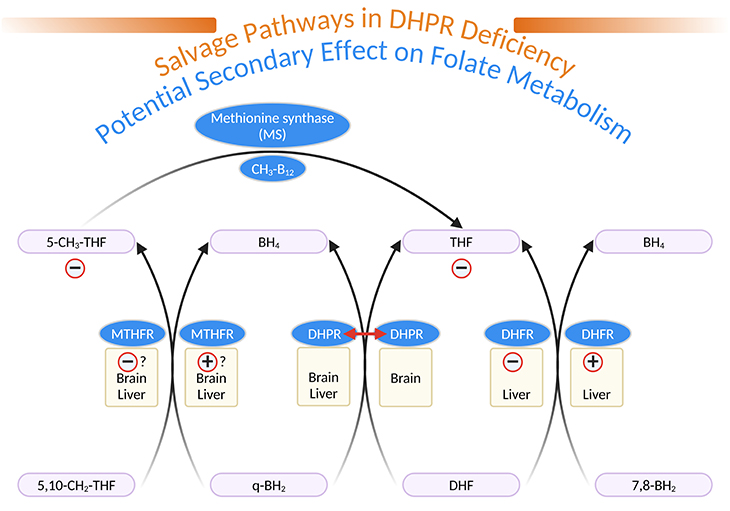
Figure 4. Salvage Pathways in Dihydropteridine Reductase (DHPR) Deficiency and Potential Secondary Effects on Folate Metabolism. [q-BH2, quinonoid dihydrobiopterin; BH4, tetrahydrobiopterin; 7,8-BH2, L-erythro-7,8-dihydrobiopterin; DHF, dihydrofolate; THF, tetrahydrofolate; 5,10-CH2-THF, 5,10-methylene-tetrahydrofolate; 5-CH3-THF, 5-methyl-tetrahydrofolate; MTHFR, 5,10-methylene-tetrahydrofolate reductase; DHPR, dihydropteridine reductase; DHFR, dihydrofolate reductase; MS, methionine synthase; CH3-B12, methylcobalamin]
The Relationship Between Biopterin and Folate Metabolism
DHPR is well represented in brain, even in the area without tyrosine or tryptophan hydroxylase activity. DHPR has been shown to catalyze the reduction of quinonoid dihydrofolate to THF (see Figure 4). DHPR deficiency results in low brain and CSF folate concentrations. DHFR, well represented in liver but not in brain can reduce 7,8-BH2 to BH4 (see Figure 4) [4].
In DHPR-deficient patients, a competition between 7,8-BH2 (derived from q-BH2 accumulated above the metabolic block) and dihydrofolate could lead to THF deficiency peripherally and secondarily in brain CSF. 5, 10-methylene-THF reductase (MTHFR), which normally catalyzes the reduction of 5,10-methylene THF to 5-methyl-THF (the essential folate derivative), present in liver and brain, could, in DHPR-deficient patients, help to reduce q-BH2 to BH4. A competition between q-BH2 and 5,10-methylene-THF, could decrease the production of 5-methyl-THF (see Figure 4).
Patients with MTHFR deficiency, who also have a severe deficiency in brain CSF 5-methyl-THF, have low levels of amine metabolites in CSF. It has been shown in a DHPR-deficient patient that some salvage of BH4 exist, likely by means of MTHFR and DHFR.
In DHFR-deficient patients, the decreased level of reduced folate in brain seems to impede the control of biogenic amine release rather than the amine synthesis. It has been demonstrated on homogenized fresh rat brain that the addition of 5-methyl-THF, hydroxocobalamin, and ascorbate increased the in vitro synthesis of biopterin.
Folinic acid (5-formyl-THF) supplementation, by providing increased amounts of reduced folate for MTHFR, peripherally and in the brain, can correct 5-methyl-THF deficiency, and thus prevent basal ganglia calcifications and delayed neurologic deterioration in DHPR-deficient patients.
5-methyl-THF seems too unstable to be used as a therapeutic agent. Not only folic acid (FA) is ineffective, but by competitive inhibition with 5-methyl-THF at the BBB level, can aggravate the neurologic status of such children.
Tetrahydrobiopterin (BH4) Deficiency in ASD
ASD is a disorder that describes individuals who have persistent deficits in: (i) social communication and social interaction; with (ii) restricted, repetitive patterns of behavior, interest, or activity. ASD represents a large spectrum of classifications and presentations, from mild to severe impairment [5].
ASD’s pathophysiology is not very well understood; however, evidence from the literature indicates that a specific modulation of the BH4 pathway affecting the activity of the monoaminergic neurons that might be downregulated in the disease. In fact, reduced levels of BH4 have been demonstrated in the blood, urine, and CSF of ASD-affected children.
Furthermore, several reports have demonstrated that a therapeutic effect of BH4 supplementation in children with ASD. BH4 daily doses of 1 to 3 mg/kg for 4 to up to 105 weeks elicited noticeable improvements in social responsiveness, communication, and cognitive abilities in over 300 mildly to severely affected ASD children. These improvements were marked in children with higher intelligence and younger than 5 years old. Remarkably, BH4 daily supplementation with 20 mg/kg induced similar effects than lower doses [6-7].
Take Home Messages
- BH4 metabolic disorder should be considered in the presence of a developmental delay, tonus abnormalities, intellectual disability, and movement disorders.
- Early treated cases suggests that clinical response may be satisfactory in BH4 deficiency.
- It is essential to evaluate both newborns presenting with hyperphenylalaninemia and patients with unknown neurological origin in terms of BH4 deficiencies.
- Deficiencies in BH4, an essential cofactor for dopamine and serotonin biosynthesis in the CNS, can result in severe neurological disorders.
- Few children with ASD may have abnormally low CSF BH4 deficiency. Most of the cases demonstrated global developmental delay without epilepsy.
Summary and Conclusions
Here we have provided an overview of the relationship between biopterin and folate metabolism and the accompanying consequences of tetrahydrobiopterin (BH4) deficiency pertaining to neuropsychiatric/neurodevelopmental conditions. As well as therapeutic benefits of BH4 replacement along with folinic acid (5-formyl-THF) to ameliorate these neurological conditions.
Most importantly, the main objective of this blog is to highlight the fact that, the significance of BH4 in ASD and other neurodevelopmental conditions should not be understated or ignored. Low CNS BH4 levels can result in devastating neurodevelopmental consequences. Because BH4 is an enhancer of synaptic release of a wide range of neurotransmitters, such as biogenic amines (serotonin, dopamine, and norepinephrine), acetylcholine, glutamate, and gamma-aminobutyric acid (GABA). Therefore, BH4 is involved in several metabolic and neurotransmitter pathways critical from CNS function and development.
Low BH4 during development could have detrimental consequences to the CNS, leading to or potentiating the neuropathology underlying ASD. The central role of BH4 in a wide range of CNS and non-CNS metabolic pathways, which are known to be dysfunctional in ASD, and one that may have a therapeutic role.
Did You Know? Folate Receptor Autoantibodies (FRAAs) may impede proper folate transport.
Folate (vitamin B9) is very important for your child’s brain development!
During pregnancy, it helps prevent neural tube defects and plays a big role in forming a normal and healthy baby’s brain and spinal cord. Folate also helps cells divide and assists in both DNA and RNA synthesis.
Emerging research suggests that the presence of FRAAs negatively impacts folate transport into the brain.
- Recent studies reveal that a large subgroup of children with autism spectrum disorder (ASD) have FRAAs.
- This suggests that a possible disruption in folate transport across the blood-cerebrospinal fluid (CSF) barrier may potentially influence ASD-linked brain development.
- Screening for the FRAAs in your child should be part of your early intervention strategies.
Is there a test for identifying Folate Receptor Autoantibodies (FRAAs)?
Yes, there is a test – The Folate Receptor Antibody Test (FRAT®) has emerged as a diagnostic tool for detecting the presence of FRAAs.
It is important to screen at an early age or as soon as possible as there may be corrective measures available. Please consult your physician for further information.
To request a test kit, click on the button below.

For information on autism monitoring, screening and testing please read our blog.
References
- Parker CE. Diseases of phenylalanine metabolism. West J Med. 1979 Oct;131(4):285-97. PMID: 388868; PMCID: PMC1271823.
https://pubmed.ncbi.nlm.nih.gov/388868/ - Smith I, Howells DW, Hyland K. Pteridines and mono-amines: relevance to neurological damage. Postgrad Med J. 1986 Feb;62(724):113-23. doi: 10.1136/pgmj.62.724.113. PMID: 3540926; PMCID: PMC2418601.
https://pubmed.ncbi.nlm.nih.gov/3540926/ - Fanet H, Capuron L, Castanon N, Calon F, Vancassel S. Tetrahydrobioterin (BH4) Pathway: From Metabolism to Neuropsychiatry. Curr Neuropharmacol. 2021;19(5):591-609. doi: 10.2174/1570159X18666200729103529. PMID: 32744952; PMCID: PMC8573752.
https://pubmed.ncbi.nlm.nih.gov/32744952/ - Banka S, Blom HJ, Walter J, Aziz M, Urquhart J, Clouthier CM, Rice GI, de Brouwer AP, Hilton E, Vassallo G, Will A, Smith DE, Smulders YM, Wevers RA, Steinfeld R, Heales S, Crow YJ, Pelletier JN, Jones S, Newman WG. Identification and characterization of an inborn error of metabolism caused by dihydrofolate reductase deficiency. Am J Hum Genet. 2011 Feb 11;88(2):216-25. doi: 10.1016/j.ajhg.2011.01.004. PMID: 21310276; PMCID: PMC3035707.
https://pubmed.ncbi.nlm.nih.gov/21310276/ - Eichwald T, da Silva LB, Staats Pires AC, Niero L, Schnorrenberger E, Filho CC, Espíndola G, Huang WL, Guillemin GJ, Abdenur JE, Latini A. Tetrahydrobiopterin: Beyond Its Traditional Role as a Cofactor. Antioxidants (Basel). 2023 May 3;12(5):1037. doi: 10.3390/antiox12051037. PMID: 37237903; PMCID: PMC10215290.
https://pubmed.ncbi.nlm.nih.gov/37237903/ - Frye RE. Tetrahydrobiopterin Deficiency in Autism Spectrum Disorder. N A J Med Sci. 2014;7(3):93-96. DOI: 10.7156/najms.2014.0703093
https://www.najms.com/index.php/najms/article/viewFile/148/140 - Frye RE, Huffman LC, Elliott GR. Tetrahydrobiopterin as a novel therapeutic intervention for autism. Neurotherapeutics. 2010 Jul;7(3):241-9. doi: 10.1016/j.nurt.2010.05.004. PMID: 20643376; PMCID: PMC2908599.
https://pubmed.ncbi.nlm.nih.gov/20643376/


