Table of Contents
- Introduction
- What are Peroxisomes?
- Metabolic and Molecular Bases of Peroxisomal Disorders
- Main Biochemical Pathways in Peroxisomes
- Fatty Acid β-Oxidation
- Fatty Acid α-Oxidation
- Zellweger Syndrome (ZS)/Neonatal Adrenoleukodystrophy (NALD)/Infantile Refsum Disease (IRD)
- Rhizomelic Chondrodysplasia Punctata (RCDP)
- X-Linked Adrenoleukodystrophy (X-ALD)
- Adult Refsum Disease (ARD)
- Diagnosis and Laboratory Findings
- Treatment of Peroxisomal Disorders
- Take Home Messages
- Conclusion
- Did You Know About Folate Receptor Autoantibodies (FRAAs) and Brain Development?
- References
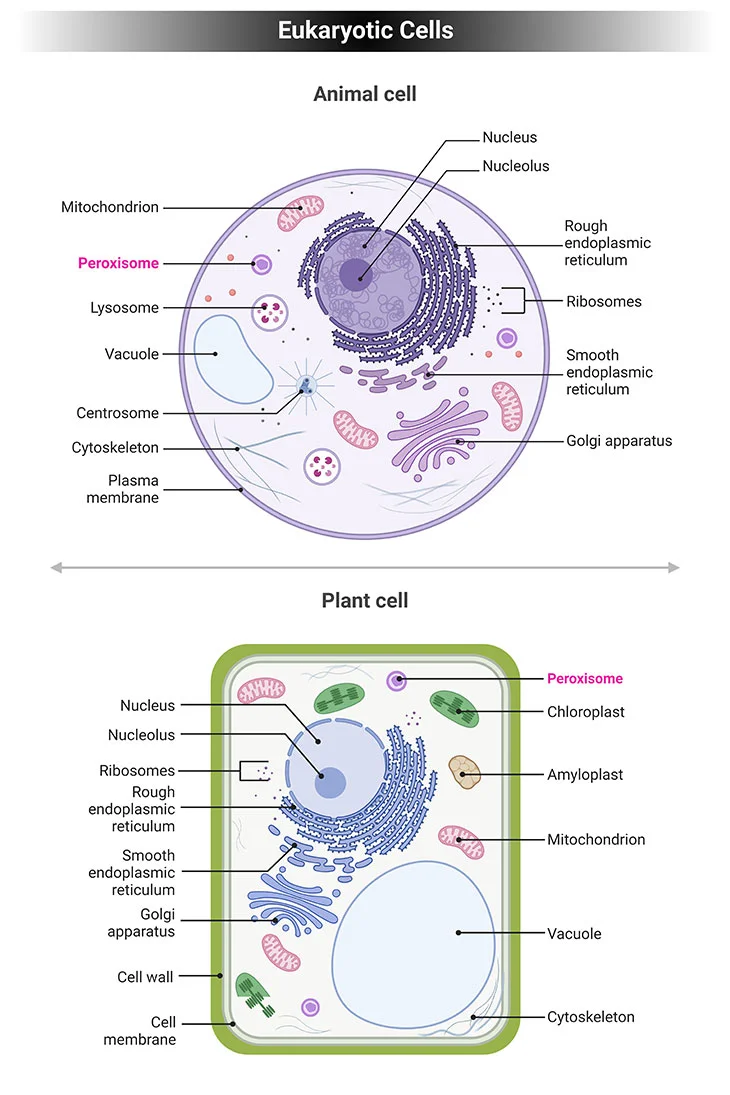
Figure 1. Eukaryotic Cells. Eukaryotic cells have specialized subcellular organelles that play distinctive roles in cellular metabolism. The membrane structure and the suborganelle content of these structures are unique in order to carry out their different roles. These organelles are essentially the nucleus, endoplasmic reticulum (ER), Golgi apparatus, mitochondria, lysosomes, and peroxisomes. Peroxisomes contain enzymes that oxidize certain biomolecules normally found in the cell, particularly fatty acids and amino acids.
Introduction

Peroxisomal disorders are a diverse group of hereditary metabolic diseases that occur as a result of partial or total malfunction of peroxisomes in the body. Hereditary disorders occurs when parents pass the defective genes that cause these disorders to their offspring (cf. previous blog entitled as ‘Developmental Delays in Infancy and Early Childhood: Phenylalanine and Tyrosine Metabolism Disorders.’).
What are peroxisomes?
Eukaryotic cells have specialized subcellular organelles that play distinctive roles in cellular metabolism. The membrane structure and the suborganelle content of these structures are unique in order to carry out their different roles. These organelles are essentially the nucleus, endoplasmic reticulum (ER), Golgi apparatus, mitochondria, lysosomes, and peroxisomes (see Figure 1).
Peroxisomes contain enzymes that oxidize certain biomolecules normally found in the cell, particularly fatty acids and amino acids. Those oxidation reactions generate hydrogen peroxide (H2O2), which is the basis of the name “peroxisome,” a type of microbody (peroxide + soma – “body’). However, hydrogen peroxide is potentially toxic to the cell since it has the ability to react with many other biomolecules. As a result, peroxisomes also contain enzymes such as catalase that convert hydrogen peroxide to water (H2O) and oxygen (O2), thus neutralizing the toxicity. In that way peroxisomes provide a safe compartment for the oxidative metabolism of certain biomolecules.
In order to better appreciate these inborn errors of metabolisms (IEMs) causing peroxisomal diseases, it is essential to gain knowledge of the function and cell biology of peroxisomes. Peroxisomes are cell organelles that consist of a protein-rich matrix surrounded by a single membrane; and were discovered by Belgian biochemist Christian de Duve in 1965, 10 years after he discovered lysosomes [1].
Current knowledge dictates that the mechanism of peroxisome biogenesis involves the following essential features (see Figure 2) [2, 3]:
- Peroxisomes are not autonomously multiplying organelles but are derived from the endoplasmic reticulum (ER);
- All peroxisomal proteins, including peroxisomal matrix and membrane proteins, are encoded by the nuclear genome and synthesized on free polyribosomes;
- The newly synthesized proteins are post-translationally imported from the cytosol into preexisting peroxisomes; and
- Import of new peroxisomal proteins into peroxisomes leads to an expansion of the size of peroxisomes, which makes them grow until a critical size is attained.
Subsequently, the peroxisomes divide into two daughter peroxisomes that can then undergo the same cycle of events.
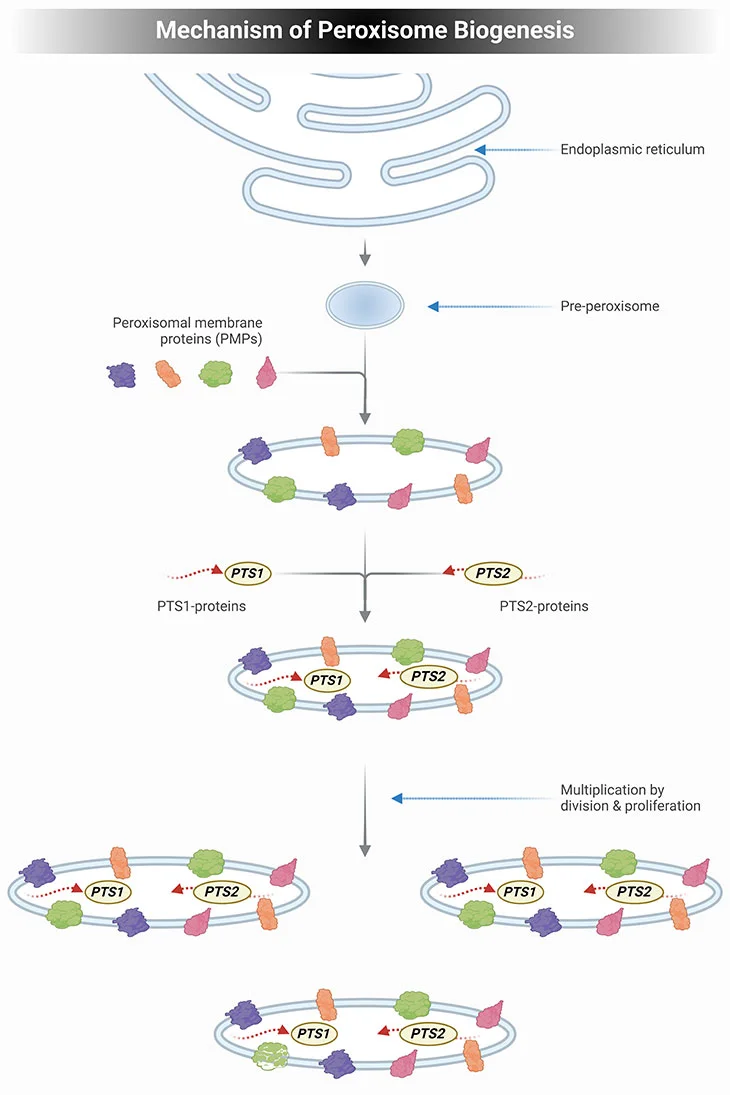
Figure 2. Mechanism of Peroxisome Biogenesis. Schematic illustrating the mechanism of peroxisome biogenesis. Current knowledge dictates that peroxisome can originate from pre-existing peroxisome by division of one peroxisome into two daughter cells but can also be derived de novo from the endoplasmic reticulum (ER) in the form of a pre-peroxisome. They subsequently develop into mature, metabolically active peroxisomes through the subsequent import of peroxisomal membrane proteins (PMPs), followed by the uptake of peroxisomal matrix proteins, containing either a PTS1 or PTS2 signal. The division of peroxisomes involves three distinct sequential steps: elongation of peroxisomes, membrane construction, and fission of peroxisomes.
Metabolic and Molecular Bases of Peroxisomal Disorders
Peroxisomes contain more than 50 different matrix enzymes that are essential for various anabolic and catabolic processes. In contrast to lysosomes, which are rich in enzymes that require an acidic environment, peroxisomal enzymes are most efficient in the oxidative surroundings.
Peroxisomes are found in all human cells, except in erythrocytes (RBCs). Depending upon their tissue type, their number in a human cell varies considerably from about 100 to 1,000. The highest numbers may be found in hepatocytes (liver cells) and kidney tubular cells, where peroxisomes are involved in the synthesis of bile acids and in detoxification processes (see Figure 3, 4).
Most crucially, in the brain, peroxisomes have a critical role in the generation of intermediate of complex lipids that are incorporated into myelin, as well as in the synthesis of docosahexaenoic acid (DHA), an omega-3 fatty acid that is a primary structural component of the cerebral cortex. Consequently, the altered function of peroxisomes in the brain invariably leads to severe neurological disorders [4].
All peroxisomal proteins, including membrane proteins and enzymes, are nuclearly encoded and synthesized in the cytosol and then imported into peroxisomes. The peroxisomal enzymes are transported into the peroxisomes by peroxins, which are encoded by PEX genes. In humans, there are at least 16 PEX genes identified, thus far. Mutations in PEX genes lead to absent or empty peroxisomes resulting in peroxisomal dysfunction. These are collectively known as peroxisomal biogenesis disorders (PBDs).
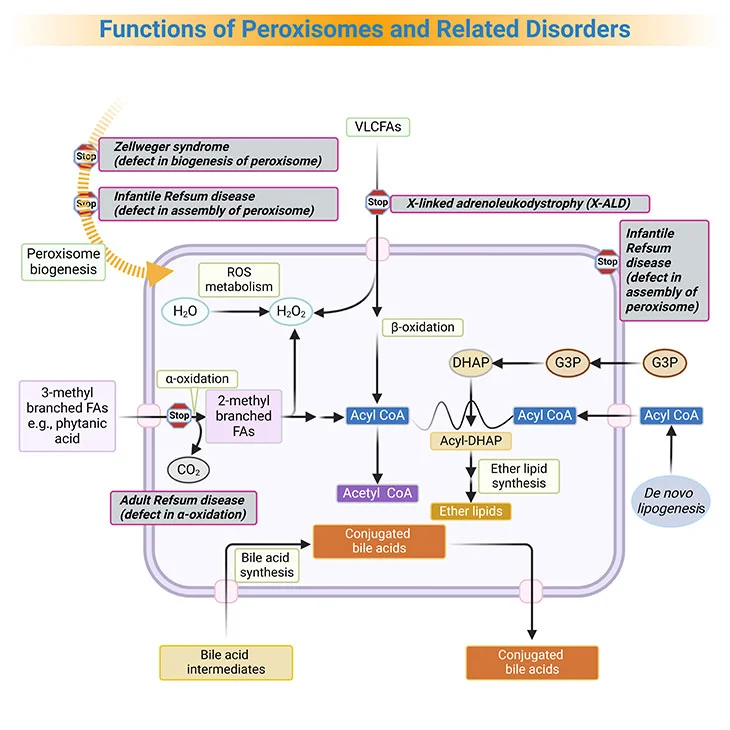
Figure 3. Functions of Peroxisomes and Related Disorders. Peroxisomes catalyze a number of essential metabolic functions. The most important, at least in human beings, are: (i) fatty acid α-oxidation; (ii) fatty acid β-oxidation; (iii) etherphospholipid biosynthesis; (iv) glyoxylate detoxification, and (v) hydrogen peroxide (H2O2) metabolism. Based on the extent of the inherited deficit, peroxisomal disorders can be divided into: (a) the peroxisomal biogenesis disorders (PBDs), where all peroxisomal functions are altered, and (b) the single peroxisomal enzyme deficiencies (PEDs), with dysfunction of a single enzyme or transporter. [FA, fatty acid; VLCFA, very long-chain fatty acids; ROS, reactive oxygen species; G3P, glycerol-3-phosphate; DHAP, dihydroxyacetone phosphate]
Based on the extent of the inherited deficit, peroxisomal disorders can be divided into [5]:
- the peroxisomal biogenesis disorders (PBDs), where all peroxisomal functions are altered, and
- the single peroxisomal enzyme deficiencies (PEDs), with dysfunction of a single enzyme or transporter.
Conditions classified as PBDs are, for example:
- Zellweger syndrome (ZS)
- Neonatal adrenoleukodystrophy (NALD)
- Infantile Refsum disease (IRD)
- Rhizomelic chondrodysplasia punctata (RCDP) type 1
Currently, ZS, NALD, and IRD are considered to be various clinical manifestations of the same biochemical disorder (also known as Zellweger syndrome spectrum, or PBD-ZSS), with ZS having the most severe disease intensity, with complete absence of peroxisomes and IRD the mildest (see Figure 3, 4).
The single peroxisomal enzyme deficiencies category is further classified according to 5 various affected peroxisomal metabolic pathways:
- Peroxisomal α-oxidation
- Peroxisomal β-oxidation
- Ether phospholipid synthesis
- Glyoxylate detoxification
- Hydrogen peroxide (H2O2) metabolism
The majority of peroxisomal disorders are inherited in an autosomal recessive manner. The exception is X-linked adrenoleukodystrophy (due to mutations in the ABCD1 transporter), which is predominantly affecting males, while females are mostly asymptomatic carriers.
In PBDs, severe damage to the central nervous system (CNS) occurs early in utero during the growth phase due to the defect in neuronal migration. Early death and severe form of mental retardation are the consequence in the majority of these cases. Those children who do survive beyond the first year of life manifest, for example:
- dysmorphic facial features,
- oculomotor dysfunction,
- motor and speech impairment (athetosis, dystonia, ataxia), and
- severe neurological developmental delays.
Main Biochemical Pathways in Peroxisomes
In order to fully grasp the laboratory findings in PBD and peroxisomal enzyme/transport defects, one should step back and learn the major biochemical pathways compartmentalized in time and space within specific subcellular organelles, especially in peroxisomes.
Peroxisomes play an important role in a number of anabolic and catabolic pathways (see Figure 3, 4).
- Catabolic pathways include, for example, beta-oxidation and alpha-oxidation of fatty acids.
- Anabolic pathways involve, for instance, biosynthesis of bile acids and plasmalogen, which can protect cells from reactive oxygen species (ROS) and are found mostly in nervous, cardiovascular, and immune systems.
- As well as the presqualene segment of the cholesterol/isoprenoid biosynthetic pathways also occurs in peroxisomes.
- Finally, as its name suggests, peroxisomes are an important organelle, especially in liver, for detoxification of various toxic molecules including alcohol through peroxidation reaction.
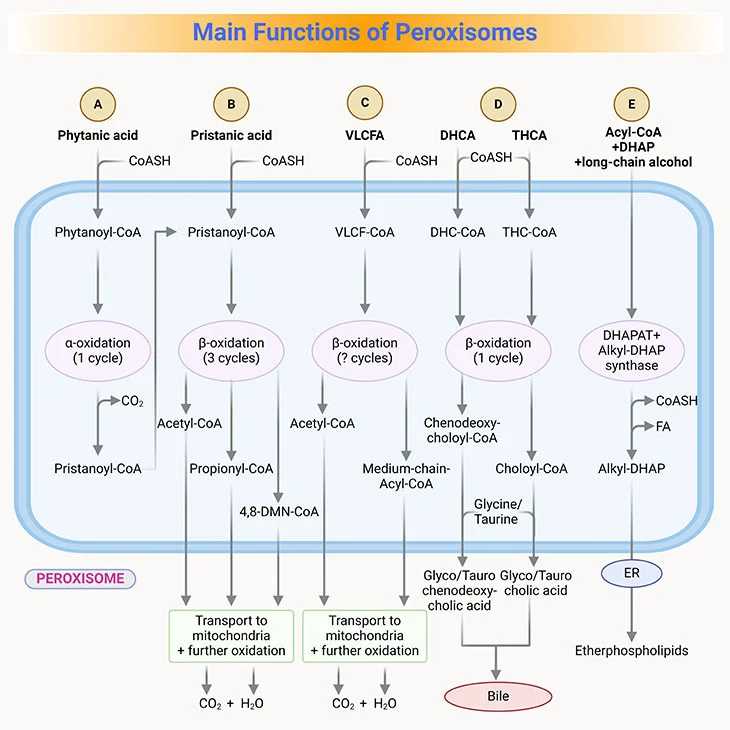
Figure 4. Main Functions of Peroxisomes in Humans. (i) Fatty acid alpha-oxidation; (ii) fatty acid beta-oxidation; (iii) etherphospholipids (plasmalogen) biosynthesis, and (iv) glyoxylate detoxification.
Fatty Acid β-Oxidation
- Peroxisomal enzymes for beta-oxidation overlap in function with those in mitochondria besides their selectivity for different lengths of fatty acids (see Figure 3, 4).
- Peroxisomal enzymes oxidize very long-chain fatty acids (VLCFA), that is those 22-26 carbons in length, to medium-chain fatty acids, which are then transported to the mitochondria for further complete oxidation to CO2 and H2O.
- β-oxidation in the peroxisomes is also important for pristanic acid metabolism, which is the product generated after α-oxidation of phytanic acid, a branched-chain amino acid commonly found in the diet though the consumption of dairy products and fats from ruminant animals, for example, goats, sheep, cattle, as well as certain fish.
- Consequently, any defect in peroxisomal β-oxidation, either due to PBD or single gene abnormalities, characteristically manifests with elevated VLCFA and phytanic acid.
- For example, X-linked adrenoleukodystrophy (X-ALD) is the most prevalent peroxisomal disorder. It affects β-oxidation of VLCFA due to the defective transport of these fatty acids into the peroxisomes because of mutations in the ABCD1 transporter gene.
- In X-ALD, the accumulation of VLCFAs may potentially be toxic to the adrenal cortex and myelin by triggering inflammatory response in the brain inducing demyelination, i.e., breakdown of myelin.
Fatty Acid α-Oxidation
- Phytanic acid, the most notorious branched-chain fatty acid, cannot be metabolized by β-oxidation as it has a methyl group at the β-position (viz., C-3). Rather, it first undergoes alpha-oxidation to remove the terminal carboxyl group as CO2 (see Figure 3, 4).
- Once pristane is formed by α-oxidation, it goes through the β-oxidation.
- The key enzyme in α-oxidation is phytanoyl-CoA hydroxylase.
- For example, in adult Refsum disease the phytanoyl-CoA hydroxylase is deficient, leading to accumulation of phytanic acid.
- Refsum disease can be successfully treated with diet by avoiding consumption of dairy-related products, and likewise fats from ruminant animals, which contains phytanic acid.
- Refsum disease if left untreated, eventually will manifest as retinitis pigmentosa, cerebellar ataxia, and neuropathy.
Zellweger Syndrome (ZS)/
Neonatal Adrenoleukodystrophy (NALD)/
Infantile Refsum Disease (IRD)
These group of disorders are 3 expressions of a disease continuum, from most (ZS) to least (IRD) severe. The responsible genetic defect occurs in 1 of at least 12 genes that are involved in peroxisomal formation or protein import, the so-called PEX gene family[2, 6].
Clinical manifestations include hypotonia, poor feeding, facial dysmorphism (distinctive facies), brain malformations, demyelination, neonatal seizures, hepatosplenomegaly, cystic kidneys, cholestasis, and hepatic dysfunction, short limbs with stippled epiphyses (chondrodysplasia punctata), cataracts, retinopathy, hearing deficit, psychomotor delay, and peripheral neuropathy.
Diagnosis is suspected when elevated blood levels of very-long-chain and branched-chain fatty acids, phytanic acid, bile acid intermediates, and pipecolic acid are detected (see Figure 5). Fasting serum analysis by gas chromatography-mass spectrometry (GC-MS) is essential. Findings, such as elevation of C26:0 and C26:1 and the increased ratios C24:C22 and C26/C22 are consistent with a defect in peroxisomal fatty acid metabolism. This is the most informative initial screen. The diagnosis of Zellweger disorder spectrum is positively established after molecular genetic testing of PEX genes. Additionally, functional testing in fibroblasts may also be used to confirm molecular and biochemical results.
There is currently no specific treatment protocols for these disorders. Management is primarily symptomatic.
Rhizomelic Chondrodysplasia Punctata (RCDP)
This disorder of peroxisomal biogenesis is caused by PEX7 gene mutations and characterized by skeletal changes that include strikingly proximal limbs (dwarfism), midface hypoplasia, frontal bossing, small nares. Vertebral clefts are also common. As well as cataracts, ichthyosis, and profound psychomotor retardation.
Diagnosis of RCDP is suspected by x-ray findings, elevation of serum phytanic acid, and low RBC plasmalogen levels; VLCFA levels are normal. Confirmation by genetic testing. There is no specific treatment of RCDP, the extent of affected vital organs and heavy disease burden significantly impact the life expectancy of patients with RCDP and cause substantial death rates even at neonatal age.
X-Linked Adrenoleukodystrophy (X-ALD)
X-ALD is the most common single peroxisomal disorder, with a minimum incidence of 1 in 21,000 males in the United States and 1 in 15,000 males in France. This disorder is caused by deficiency of the peroxisomal membrane transporter ALDP, which is coded for by the gene ABCD1. This is an X-linked gene and thus the disorder manifests primarily in males. So far, greater than 900 mutations have been identified.
The cerebral form affects 40% of patients. Onset of the disease occurs between age 4 years and 8 years, and symptoms of attention deficit progress over time to severe behavioral problems, dementia, as well as vision, hearing, and motor deficits, causing total disability and death 2 to 3 years after diagnosis. Milder adolescent and adult forms have also been described.
In general, about 45% of patients have a milder form called adrenomyeloneuropathy (AMN); onset occurs in the 20s or 30s, with progressive paraparesis, and sphincter and sexual disturbance. About one third of these patients also eventually develop cerebral symptoms. Patients with any form may also develop adrenal insufficiency; about 15% have isolated Addison disease without neurologic involvement.
Stem cell or bone marrow transplantation may help stabilize symptoms in some cases. Adrenal steroid replacement is needed for patients with adrenal insufficiency. Dietary supplement with a 4:1 mixture of glyceryl trioleate and glyceryl trierucate (Lorenzo’s oil) can normalize plasma VLCFA levels but does not appear to stop neurologic degeneration in symptomatic patients. However, if given to boys before symptom onset, it may slow disease progression; the exact benefit has not been determined. Gene therapy trials are currently underway and have shown some preliminary success.
Adult Refsum Disease (ARD)
Genetic deficiency of a single peroxisomal enzyme, phytanoyl-CoA hydroxylase, which catalyzes metabolism of phytanic acid (a common dietary plant component, derived by bacterial fermentation of green plants or algae), causes phytanic acid accumulation.
Clinical manifestation include progressive peripheral neuropathy, sensorineural deafness (hearing deficit), impaired vision caused by retinitis pigmentosa, anosmia, ichthyosis (dry, scaly or thickened skin), and cardiomyopathy and conduction defects. Onset of the disease is usually in the twenties.
Diagnosis of ARD is established by elevation of serum phytanic acid and decreased levels of pristanic acid (phytanic acid elevation is accompanied by pristanic acid elevation in several other peroxisomal disorders). Management of ARD is dietary restriction of phytanic acid (< 10 mg/day), which can be effective in prevention and/or delaying symptoms when initiated before symptoms onset.
Diagnosis and Laboratory Findings
Laboratory tests include targeted biochemical analysis of various metabolites in the blood and urine (see Figure 5) [7, 8].
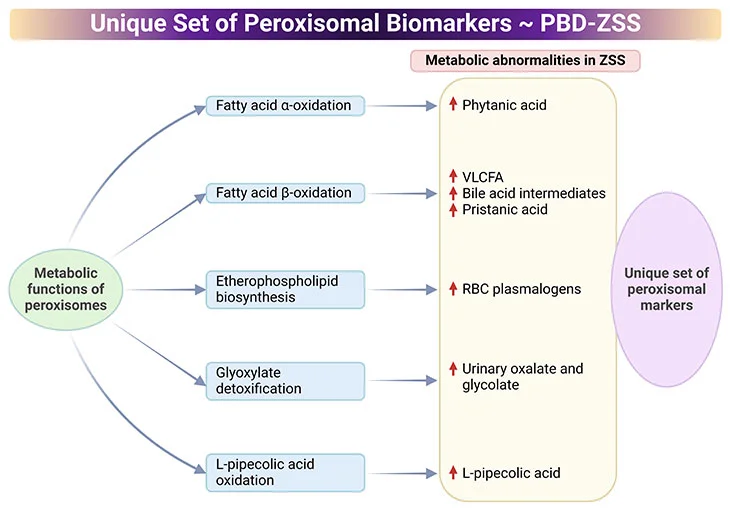
Figure 5. Distinctive Set of Peroxisomal biomarkers. Peroxisomal biomarkers used in the identification of peroxisomal biogenesis disorders – Zellweger syndrome spectrum (PBD-ZSS). Laboratory tests include targeted biochemical analysis of various metabolites in the blood and urine.
-
- Common assays in plasma are measurements of very-long-chain fatty acids (VLCFAs), dihydroxycholetanoic acid (DHCA), and trihydroxycholetanoic acid (THCA), all intermediates of the bile acid metabolism.
- Levels of branched-chain fatty acids, (viz., phytanic, pristanic, and pipecolic acid) are also routinely analyzed.
- Excessive levels of VLCFA cerotic acid (C26:0) and the high ratio of C26:0 to docosanoic acid (C22:0) indicate the alteration of the peroxisomal fatty acid metabolism.
- Pathological laboratory findings indicative of peroxisomal disorder include high plasma levels of DHCA, THCA, as well as pristanic, pipecolic, and phytanic acid.
- Erythrocytes are used for the measurement of plasmalogens, a unique class of membrane ether phospholipids. Levels of C16 and C18 plasmalogen are typically low in peroxisomal disorders.
- Urine samples are suitable for the measurement of the levels of bile acids and oxalic acids.
Finally, dried blood spot (DBS) is a convenient specimen used in newborn/neonatal screening for NALD. DBSs are typically analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS), where increased levels of C26:0-lysophosphatidylcholine are indicative of NALD. The advantage of this test is also in the detection of other disorders within PBD-ZSS that are accompanied by elevated VLCFA levels.
Treatment of Peroxisomal Disorders
For decades, management of peroxisomal disorders has been mostly symptomatic and palliative. Because these conditions often affect multiple organs and systems, a comprehensive multidisciplinary approach and personalized therapy are essential for improving the quality of the life of affected individuals.
Over recent years, gene therapy for peroxisomal diseases is being rapidly developed. Considering the fact that the majority of peroxisomal disorders impact brain development, novel gene therapies are predominantly aiming to alleviate brain damage and its associated neurological consequences.
There are two strategic types of gene therapy for brain involvement in peroxisomal disorders, for instance:
- the direct transfer of a therapeutic gene into brain cells, and
- hematopoietic stem cell-targeted gene therapy.
The rationale for the latter approach is that brain microglia is derived from hematopoietic cells. Consequently, gene-corrected hematopoietic cells migrate into the brain and their differentiation into microglia could potentially reduce the inflammation; and yield a clinical benefit in patients with peroxisomal disorders. However, these approaches are still in the clinical trial phase, very encouraging results have been obtained.
Take Home Messages
- Peroxisomes are semiautonomous subcellular organelles that can form de novo and/or by growth and division of preexisting organelles.
- Peroxisomal disorders are hereditary metabolic disorders, may occur either by a single gene defect or a biogenesis disorder referring to ineffective assembly of the peroxisome itself.
- Peroxisomes have both anabolic and catabolic metabolic functions. They are β-oxidation of VLCFAs and removal of unwanted macromolecular waste products; as well as the biosynthesis of cholesterol, bile acids, platelet-activating factors, and plasmalogens, respectively.
- Proper functioning of peroxisomes in metabolism requires concerted interaction with other subcellular organelles including mitochondria, lysosomes, endoplasmic reticulum, lipid droplets, and the cytosol.
- In the early neonatal period, a child with failure to thrive, hypotonia, dysmorphic facial features, brain malformations, hepatosplenomegaly, elevated transaminases, and lipid abnormalities should be suspected of having a peroxisomal disorder.
- Zellweger syndrome represents the most common occurring of the peroxisomal disorders.
- PEX gene analysis aids in establishing a diagnosis of peroxisomal spectrum disorders.
- One of the more recently identified functions of peroxisomes involves their role in immune function and viral infections.
Conclusion
Here we have presented a succinct overview of peroxisomal disorders pertaining to developmental delays in infancy and early childhood. Peroxisomal disorders are a heterogeneous group of inborn errors of metabolism caused by impairment in the biogenesis of peroxisomes or one of their metabolic functions. In most cases, this results in neurologic dysfunction of varying extent. Until now, unfortunately, only supportive care and palliative care are available. In recent years, gene therapy for peroxisomal diseases is being rapidly developed, very encouraging results have been reported!
Did You Know? Folate Receptor Autoantibodies (FRAAs) may impede proper folate transport.
Folate (vitamin B9) is very important for your child’s brain development!
During pregnancy, it helps prevent neural tube defects and plays a big role in forming a normal and healthy baby’s brain and spinal cord. Folate also helps cells divide and assists in both DNA and RNA synthesis.
Emerging research suggests that the presence of FRAAs negatively impacts folate transport into the brain.
- Recent studies reveal that a large subgroup of children with autism spectrum disorder (ASD) have FRAAs.
- This suggests that a possible disruption in folate transport across the blood-cerebrospinal fluid (CSF) barrier may potentially influence ASD-linked brain development.
- Screening for the FRAAs in your child should be part of your early intervention strategies.
Is there a test for identifying Folate Receptor Autoantibodies (FRAAs)?
Yes, there is a test – The Folate Receptor Antibody Test (FRAT®) has emerged as a diagnostic tool for detecting the presence of FRAAs.
It is important to screen at an early age or as soon as possible as there may be corrective measures available. Please consult your physician for further information.
To request a test kit, click on the button below.

For information on autism monitoring, screening and testing please read our blog.
References
- Sabatini DD, Adesnik M. Christian de Duve: Explorer of the cell who discovered new organelles by using a centrifuge. Proc Natl Acad Sci U S A. 2013 Aug 13;110(33):13234-5. doi: 10.1073/pnas.1312084110. Epub 2013 Aug 7. PMID: 23924611; PMCID: PMC3746853.
https://pubmed.ncbi.nlm.nih.gov/23924611/ - Wanders RJA, Baes M, Ribeiro D, Ferdinandusse S, Waterham HR. The physiological functions of human peroxisomes. Physiol Rev. 2023 Jan 1;103(1):957-1024. doi: 10.1152/physrev.00051.2021. Epub 2022 Aug 11. PMID: 35951481.
https://pubmed.ncbi.nlm.nih.gov/35951481/ - Waterham HR, Ferdinandusse S, Wanders RJ. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta. 2016 May;1863(5):922-33. doi: 10.1016/j.bbamcr.2015.11.015. Epub 2015 Nov 22. PMID: 26611709.
https://pubmed.ncbi.nlm.nih.gov/26611709/ - Berger J, Dorninger F, Forss-Petter S, Kunze M. Peroxisomes in brain development and function. Biochim Biophys Acta. 2016 May;1863(5):934-55. doi: 10.1016/j.bbamcr.2015.12.005. Epub 2015 Dec 11. PMID: 26686055; PMCID: PMC4880039.
https://pubmed.ncbi.nlm.nih.gov/26686055/ - Steinberg SJ, Raymond GV, Braverman NE, Moser AB. Zellweger Spectrum Disorder. 2003 Dec 12 [updated 2020 Oct 29]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024. PMID: 20301621.
https://pubmed.ncbi.nlm.nih.gov/20301621/ - Waterham HR, Ebberink MS. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim Biophys Acta. 2012 Sep;1822(9):1430-41. doi: 10.1016/j.bbadis.2012.04.006. Epub 2012 Apr 25. PMID: 22871920.
https://pubmed.ncbi.nlm.nih.gov/22871920/ - De Biase I, Tortorelli S, Kratz L, J Steinberg S, Cusmano-Ozog K, Braverman N; ACMG Laboratory Quality Assurance Committee. Laboratory diagnosis of disorders of peroxisomal biogenesis and function: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2020 Apr;22(4):686-697. doi: 10.1038/s41436-019-0713-9. Epub 2019 Dec 11. PMID: 31822849.
https://pubmed.ncbi.nlm.nih.gov/31822849/ - Dorninger F, Forss-Petter S, Berger J. From peroxisomal disorders to common neurodegenerative diseases – the role of ether phospholipids in the nervous system. FEBS Lett. 2017 Sep;591(18):2761-2788. doi: 10.1002/1873-3468.12788. Epub 2017 Sep 7. PMID: 28796901; PMCID: PMC5856336.
https://pubmed.ncbi.nlm.nih.gov/28796901/


