Table of Contents
- Introduction
- Global Developmental Delay (GDD)
- Overview of Amino Acid Metabolism Disorders
- Clinical Significance
- Disorders of Branched-Chain Amino and Organic Acid Metabolism
- Catabolism of Branched-Chain Amino Acids (BCAAs)
- Biochemical Diagnosis of Organic Acidemias
- Treatment of Organic Acidemias
- Take Home Messages
- Conclusion
- Did You Know About Folate Receptor Autoantibodies (FRAAs) and Brain Development?
- References
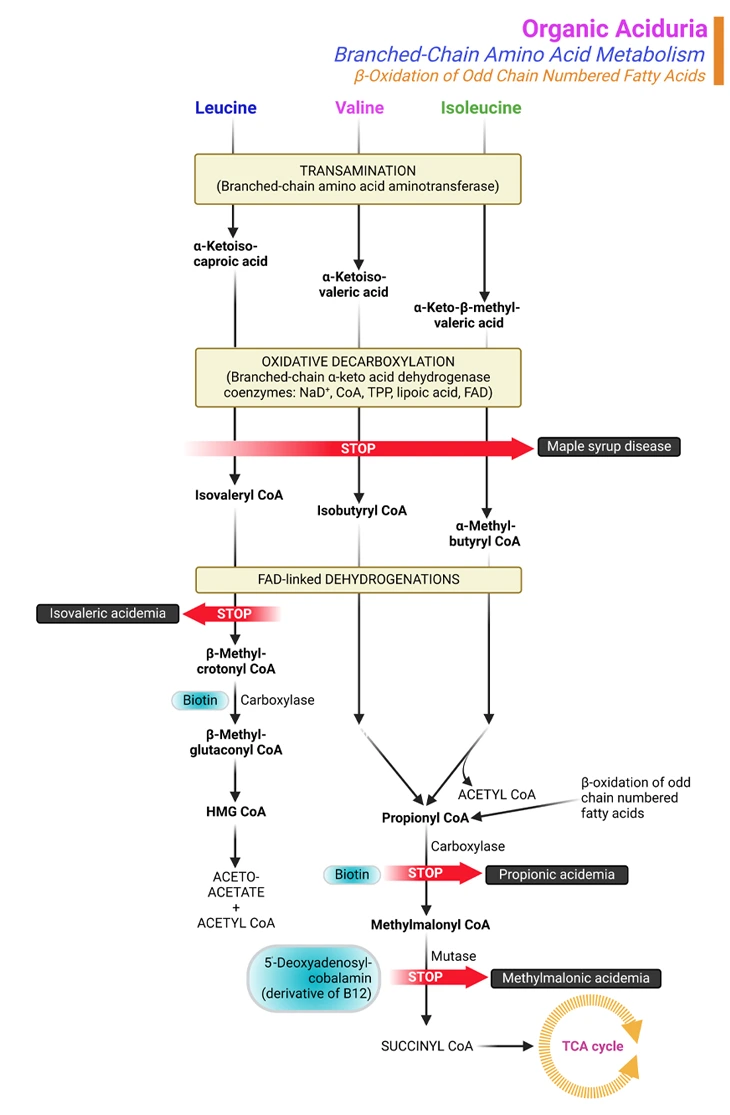
Figure 1. Organic Aciduria. Organic aciduria due to branched-chain amino acids (BCAAs) catabolism and β-oxidation of odd-chain fatty acids. The most frequent inborn errors affecting the BCAA catabolic pathway are maple syrup urine disease (MSUD) and the isovaleric, propionic, and methylmalonic acidemias (or acidurias). These 4 disorders can present with neonatal metabolic collapse, as a late-onset acute intermittent form, or as a chronic progressive form. Many other rarer defects have also been described in this catabolic pathway. The biochemical diagnosis of organic acidemias and treatment monitoring involve analysis of: (i) metabolites, (ii) enzymatic activity, and/or (iii) DNA sequence.
Introduction

Global Developmental Delay (GDD)
Children aged less than 6 years are considered to have global developmental delay (GDD) if they perform more than 2 standard deviations below age-matched peers in 2 or more aspects of development. GDD affects an estimated 1 to 3% of children, many of whom will demonstrate intellectual disability (ID) [1].
GDD/ID is not a specific diagnosis. GDD/ID is rather a description of certain conditions, in which intellectual functioning or at least two areas of adaptive performance show significant limitations when compared to the expected level for age. The adaptive areas include: (i) gross or fine motor skills, (ii) speech or language, (iii) cognition, (iv) social or personal, and (v) activities related to daily living. A significant delay is described as two or more standard deviations from the age and cultural norms (see Box-1) [2].
The prevalence of GDD/ID is approximately 3% of preschool children with some studies suggesting up to 16% of preschool children referred to pediatric/neurology clinics have some developmental problems. Accordingly, developmental delay/intellectual disability is a common medical condition, and determining the etiology can be challenging (see Box-2) [3]. (cf. previous blog entitled as ‘Developmental Delays in Infancy and Early Childhood: Metabolism of Homocysteine’ )

Overview of Amino Acid Metabolism Disorders
Amino acid metabolic disorders are hereditary metabolic disorders; collectively affect approximately 1 in 8000 newborns and affect both males and females. Hereditary disorders occur when parents pass the defective genes that cause these disorders on to their children.
In most hereditary metabolic disorders, both parents of the affected child carry a copy of the abnormal gene. Because typically two copies of the abnormal gene are necessary for the disorder to occur, generally neither parent has the disorder (see Figure 2). However, some hereditary metabolic disorders are X-linked, which indicates only one copy of the abnormal gene can cause the disorders in boys.
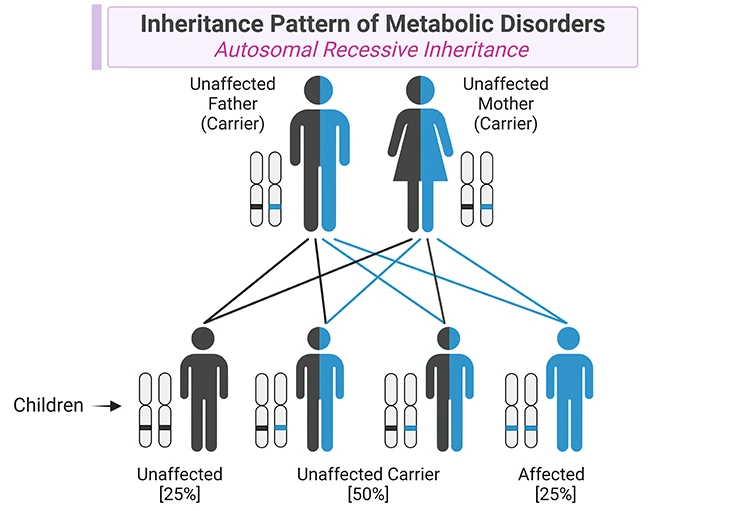
Figure 2. Inheritance Pattern of Metabolic Disorders. Metabolic disorders are caused by mutations in genes that code for specific enzymes or transporters involved in metabolic pathways. Most of metabolic disorders have autosomal recessive inheritance, in which affected individuals have a mutation in both alleles encoding for a specific enzyme or transporter. The parents of children with one of these metabolic conditions are carriers of the condition, i.e., they carry one normal allele and one mutant allele, and they do not show clinical manifestations of the disorder. In each singleton pregnancy when both parents are carriers, there is: (i) a 25% (1 in 4) chance that the child is affected; (ii) a 50% (2 in 4) chance that the child is a carrier like the parents; and (iii) a 25% (1 in 4) chance that the child has two normal alleles. Most importantly, girls are affected as often as boys.
Almost all inborn errors are transmitted as autosomal recessive traits; and result from a lack of a specific enzyme in the metabolic pathway of an amino acid, leading either to the accumulation of: (i) the parent amino acid, (ii) its by-products, or (iii) the catabolic products (organic acids), depending on the location of the enzyme block (see Figure 3) [4].
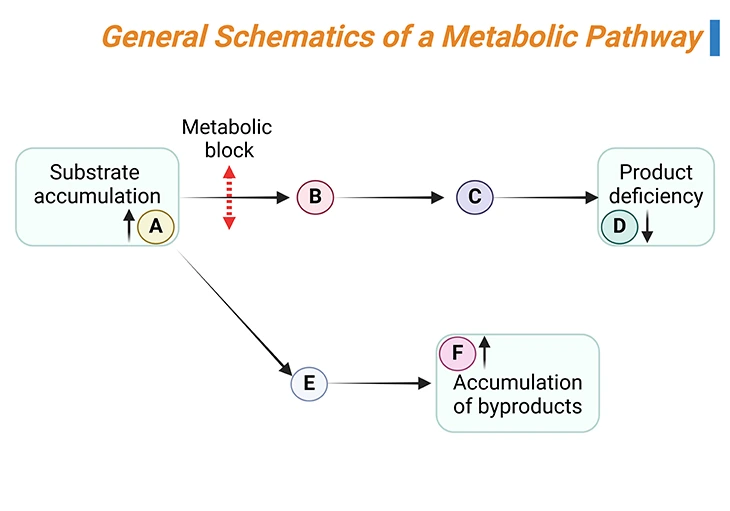
Figure 3. General Schematics of a Metabolic Pathway. The substrate A is converted by a series of reactions into product D. If one of the enzymes (rightwards black arrows) is defective (metabolic block, up down red arrow), the substrate of the reaction will accumulate (A in this case) and can enter alternative pathways of metabolism, leading to the formation of by-products (E and F in this case). At the same time, the concentration of the product of the reaction (D) will decrease.
Disorders of amino acid metabolism are divided into two groups:
- Aminoacidopathies (in which the parent amino acid accumulates in excess in blood and spills over into urine), and
- Organic acidemias (in which intermediary products [organic acids] in the catabolic pathway of certain amino acids accumulate).
An example of an aminoacidopathy is phenylketonuria (PKU), a disorder of the phenylalanine metabolism is caused in the majority of cases by deficiency of phenylalanine hydroxylase, the enzyme responsible for the conversion of phenyl alanine to tyrosine. Other examples include:
- Tyrosinemia type I
- Homocystinuria
- Maple syrup urine disease
An example of an organic acidemia is glutaric acidemia type I, a disorder of lysine and tryptophan metabolism. Other examples include:
- Isovaleric acidemia
- Methylmalonic acidemia
- Propionic acidemia
Amino acids are the building blocks of proteins and have numerous functions in the body. The body makes some of the amino acids it needs and gets others from food. Hereditary disorders of amino acid processing (metabolism) can result from defects either in the breakdown of amino acids or in the body’s ability to get amino acids into cells.
Because these disorders cause symptoms early in life, ‘newborns are routinely screened’ for several common amino acid disorders. In the United States, newborns are routinely screened, for example:
- Phenylketonuria
- Tyrosinemia
- Homocystinuria
- Maple syrup urine disease
Newborns also are screened for a number of other inherited disorders, but screening varies from state to state. Current recommendations for the ‘uniform screening panel’ for core conditions [visit: Recommended Uniform Screening Panel | HRSA; List of conditions: SACHDNC Recommended Uniform Screening Panel1 (hrsa.gov)].
Clinical Significance
The clinical manifestation of the organic acidemias vary from no observable clinical consequence to neonatal mortality. Conditions, such as: (a) developmental retardation, (b) seizures, (c) alterations in sensorium, (d) failure to thrive, or (f) behavioral disturbances, occur in more than half of the disorders.
Metabolic ketoacidosis (buildup of acid in the blood), often accompanied by hyperammonemia (increase in ammonia in the blood), is a frequent finding in organic acidemias. The compounds(s) accumulated depend on the:
- Site of the enzymatic block,
- Reversibility of the reaction proximal to the lesion, and
- Availability of alternative pathways of metabolic ‘runoff’.
Disorders of Branched-Chain Amino and Organic Acid Metabolism
Branched-chain amino acids are called ‘branched-chain’ because of their chemical structure. Leucine, isoleucine, and valine are the BCAAs that are the building blocks of proteins in the body. If these amino acids are not properly metabolized, they and their toxic by-products build up in the blood and urine, causing certain disorders (see Figure 1) [5].
Organic acidurias also known as ‘organic acidemias,’ are a set of disorders characterized by increased excretion of nonamino organic acids in urine. There are several organic acidurias, but they can be broadly categorized into five groups: viz.,
- Branched-chain organic acidemias,
- Fatty acid oxidation defects,
- Multiple carboxylase deficiencies,
- Glutaric acidurias, and
- Disorders of energy metabolism.
Most of the organic acidurias are due to the reduced activity of mitochondrial enzymes metabolizing CoA-activated carboxylic acids that are derived from amino acid catabolism.
Carnitine is a carrier molecule that shuttles long-chain fatty acyl-CoA in and out of mitochondria. However, when acyl-CoA molecules accumulate due to defective β-oxidation in the mitochondria, independent of their lengths, they are esterified with free carnitine resulting in the formation of acyl carnitines that are a hallmark to diagnose each of the organic acidurias.
The most frequent organic acidurias are: (i) isovaleric aciduria (ii) propionic aciduria, and (iii) methylmalonic aciduria (see Figure 1).
Propionic and methylmalonic acidurias are disorders of propionyl CoA metabolism from either isoleucine or valine, two of the three branched-chain amino acids and odd-chain fatty acid metabolism. Isovaleric aciduria is a disorder of leucine metabolism, the other branched-chain amino acid.
The organic acidurias are inherited in an autosomal recessive manner (See Figure 2), and they are the most common acutely life-threatening inborn errors of metabolism (IEMs). They can be diagnosed by accumulation of organic acids as acylcarnitines upstream of the genetic block. They are also often characterized with, for example:
- Elevated glycine levels,
- Ketoacidosis,
- Hyperammonemia, and
- Lactic acidosis.
Hyperglycinemia arises due to the inhibition of a glycine cleavage enzyme in the liver. The mechanism of hyperammonemia is due to the derangement of the mitochondrial energy metabolism by organic acids that are also found in the mitochondria. The urea cycle depends on mitochondria in two ways:
- Several urea cycle enzymes reside in mitochondria, and it requires ATP generated in mitochondria by oxidative phosphorylation to power the urea cycle.
- Impairment of oxidative phosphorylation and the pyruvate dehydrogenase complex results in reduced ATP synthesis and increased dependence on anaerobic glycolysis, promoting lactic acidosis.
The clinical presentation of these disorders is due to the effects of these toxic molecules on the: (a) brain, (b) liver, (c) kidney, (d) pancreas, (e) retina, and (f) other organs.
Catabolism of Branched-Chain Amino Acids (BCAAs)
The 3 essential BCAAs, viz., leucine, isoleucine, and valine, encompass about 25% of human protein. They are metabolized in mitochondria. The first 2 catabolic steps are common to the 3 BCAAs (see Figure 1). The first chemical reaction, which occurs primarily in muscle, essentially involves reversible transamination to 2-oxo (or keto) acids and is followed by oxidative decarboxylation to coenzyme A (CoA) derivatives by branched-chain oxo (or keto) acid dehydrogenase (BCKD). BCKD is similar in structure to pyruvate dehydrogenase (and shares a common subunit), is also highly regulated by a kinase/phosphatase system; and plays a key role in nitrogen metabolism.
Subsequently, the degradative pathway of BCAAs diverges. Leucine is catabolized to acetoacetate (making it a ketogenic amino acid) and acetyl-CoA, which enters the tricarboxylic acid cycle (TCA/citric acid cycle/Krebs cycle). The final step in the catabolism of isoleucine involves cleavage into acetyl-CoA and propionyl-CoA, the latter of which enters the TCA cycle via conversion to succinyl-CoA. Valine is also ultimately catabolized to propionyl-CoA (see Figure 1).
The most frequent inborn errors affecting the BCAA catabolic pathway are maple syrup urine disease (MSUD) and the isovaleric, propionic, and methylmalonic acidemias (or acidurias). These 4 disorders can present with neonatal metabolic collapse, as a late-onset acute intermittent form, or as a chronic progressive form. Many other rarer defects have also been described in this catabolic pathway [6, 7].
1. Maple Syrup Urine Disease (MUSD)
Mape syrup urine disease (MUSD) is an autosomal recessive disorder with an incidence of approximately 1:200,000 live births. Children with MSUD are unable to metabolize leucine, isoleucine, and valine. It is caused by a deficiency of the branched-chain α-keto acid dehydrogenase complex (BCKDC), and thiamine deficiency reduces the activity of branched-chain amino acid dehydrogenase; as a result, leading to a build-up of the BCAAs leucine, isoleucine, and valine and their toxic ketoacids in the blood and urine. These by-products also cause body fluids and substances, such as urine, sweat, and earwax, to smell like maple syrup. This disease is most common among Mennonite families.
There are many forms of maple syrup urine disease, depending on the gene affected and severity of the mutations. In the most severe form, infants have poor feeding and vomiting during the first week of life and develop neurologic abnormalities, including seizures and coma, during the first days of life and eventually can die within days to weeks if untreated. In the milder forms, children initially appear normal, but during infection, surgery, or other physical stress, they can develop symptoms such as vomiting, staggering, confusion, and coma.
In United States, nearly every state has required that all newborns be screened for MSUD with a blood test. Routine laboratory work reveals the presence of ketones in urines. Diagnosis is established by measuring blood amino acids and looking for elevated levels BCAAs and the presence of the alloisoleucine, which is characteristic of this disease. The diagnosis is confirmed by genetic testing.
The mainstays of treatment include special diets that is low in BCAAs and include supplementation with: (1) high-dose thiamine (injections of vitamin B1), and in many patients, (2) valine and (3) isoleucine. As well as an emergency plan in place for how to handle a sudden attack that precipitates the metabolic crisis (build-up of toxic substances in the blood and low blood sugar level). Sudden attacks are most often triggered by common infections.
2. Isovaleric Acidemia
Isovaleric acidemia is a rare autosomal recessive metabolic disorder that disrupts or prevents normal metabolism of the BCAA leucine. When leucine is not properly metabolized, harmful levels of isovaleric acid build up in the body. In isovaleric acidemia, the enzyme that is responsible for breaking down leucine, called isovaleryl CoA dehydrogenase, is not present or not working properly.
A characteristic feature of isovaleric acidemia is a distinctive odor of sweaty feet. This odor is caused by the buildup of a compound called isovaleric acid in affected individuals.
There are two forms of isovaleric acidemia. One form manifests during the first few days of life, and the other form manifests several months or years after birth. Signs and symptoms that occur in the first few days of life include poor feeding, vomiting, breathing problems, seizures, and lack of energy that can progress to coma as infants develop a buildup of acid in the blood (metabolic acidosis), low blood sugar (hypoglycemia), and an increase in ammonia in the blood (hyperammonemia). In the other form, the signs and symptoms of the disorder appear during childhood and may come and go over time. And are similar to the symptoms of the form that manifests earlier but are less severe. Quite often, they are triggered by an infection.
Physicians diagnose isovaleric acidemia by doing tests of blood and urine to detect elevated levels of isovaleric acid.
Treatment consists of dietary protein restriction, specifically consumption of leucine. During acute episodes, glycine is sometimes given, which conjugates with isovalerate forming isovalerylglycine (which help the body get rid of the excess acid), or carnitine which has similar effect.
3. Propionic Acidemia
Propionic aciduria is caused by autosomal recessively inherited deficiency of propionyl CoA carboxylase (PCC), the first step in the propionate metabolism, in which propionyl CoA is converted to methylmalonyl-CoA and then channeled into the TCA cycle.
When propionyl CoA carboxylase is not functional, harmful levels of propionic acid buildup in the body.
Propionic acidemia is found in about 1 in 35,000 live births in the United States. In most affected infants, symptoms begin to manifest in the first days or weeks after birth and include poor feeding, vomiting, and breathing problems as the infants develop a buildup of acid in the blood (metabolic acidosis), low blood sugar (hypoglycemia), and an increase in ammonia in the blood (hyperammonemia). Seizures or coma may occur. Stressors, such as fasting, fever, or infection, may trigger an acute attack. Most importantly, children who survive this life-threatening disorder may have intellectual disability and neurologic abnormalities, as well as heart and kidney problems.
When suspected, physicians diagnose propionic acidemia by doing tests of blood and urine to detect elevated levels of propionic acid. Eventually, the diagnosis is confirmed by measuring levels of propionyl CoA carboxylase in leukocytes (white blood cells) or other tissue cells and/or genetic testing.
Management of children with propionic acidemia should be started as soon as possible on a low protein diet. The patient should also receive carnitine supplement and should be given antibiotics consistently in order to remove the intestinal propiogenic flora (because bacteria in their intestines can cause propionic acid to build up). Above all, care providers should have an emergency plan in place for how to handle sudden acute attack because it may result in a buildup of toxic substances in the blood and low blood sugar, leading to metabolic crisis.
4. Methylmalonic Acidemia
Methylmalonic acidemia is an autosomal recessive metabolic disorder (1 in 48,000 births) that disrupts normal amino acid metabolism, where methylmalonyl-CoA (coenzyme A) is converted into succinyl-CoA by the enzyme methylmalonyl-CoA mutase.
Vitamin B12 is also needed for the conversion of methylmalonyl-CoA to succinyl-CoA. Mutations leading to defects in vitamin B12 metabolism or in its transport frequently result in the development of methylmalonic acidemia.
When a certain enzyme is not functional, harmful levels of methylmalonic acid build up in the body. Moreover, this disorder may be caused by deficiency of vitamin B12 (cobalamin). The age at which symptoms start, symptoms, and treatment are pretty similar to those of propionic acidemia except that physicians may give supplements of vitamins B12 instead of biotin.
Biochemical Diagnosis of Organic Acidemias
The biochemical diagnosis of organic acidemias and treatment monitoring involve analysis of:
- Metabolites,
- Enzymatic activity, and/or
- DNA sequence.
All of above 4 disorders can be diagnosed by identifying acylcarnitines and organic compounds in plasma and urines by:
- Gas chromatography (CG)-mass spectrometry (MS), or
- Tandem mass spectrometry (MS/MS)
Treatment of Organic Acidemias
In a similar way to the treatment regimens for aminoacidopathies, therapy for organic acidemias consist of:
- special diets restricting the compounds (usually amino acids) that result in the formation of the abnormal organic acid,
- supplementation with vitamins specific for each disorder.
- carnitine supplements, and
- sometimes avoidance of fasting.
For some of these conditions, aggressive therapy of illnesses with intravenous fluids containing glucose is essential to avoid catabolism that aggregates clinical symptoms.
Take Home Messages
- Disorders of amino acid metabolism result from impaired activity of an enzyme involved in the metabolic pathway of an amino acid.
- The impaired enzyme activity leads to the accumulation of the parent amino acid, its by-products, or its catabolic products, for e.g., organic acids.
- In aminoacidopathies the parent amino acid accumulates in the blood; in organic acidemias the intermediary products in the catabolic pathway of amino acids accumulate and are excreted in the urine.
- Maple syrup urine disease, phenylketonuria, tyrosinemia, and homocystinuria are examples of aminoacidopathies.
- Propionic acidemia, isovaleric acidemia, methyl melonic acidemia, and glutaric acidemia type I are examples of classical organic acidemias.
- The treatment consists of a special diet that restricts the offending amino acid(s) and includes supplements of vitamins and cofactors, including carnitine for organic acidemias. Fasting should also be avoided at all times.
Conclusion
In this blog, we have presented an overview of organic aciduria due to branched chain amino acids catabolism and its associated neurological manifestations. Organic acidurias are inherited neurometabolic diseases predominantly caused by deficiencies in enzymes involved in amino acid degradation, which results in accumulation of organic acids in the brain and other tissues.
Typically, in classical cases, disease presentation occurs in infancy, although late-onset variants can emerge during childhood or adulthood. Affected children essentially manifest with acute encephalopathy with life-threatening systemic manifestations (as in the case of classical organic aciduria); or progressive neurological symptoms (as in the case of cerebral organic aciduria), leading to permanent cerebral abnormalities.
Some organic acidurias are amenable to treatment, and early diagnosis and treatment implementation have significantly decreased the mortality and overall morbidity from organic acidurias. Nevertheless, long-term irreversible cerebral and systemic complications or sequelae are frequent because the therapeutic options are currently very limited.
Did You Know? Folate Receptor Autoantibodies (FRAAs) may impede proper folate transport.
Folate (vitamin B9) is very important for your child’s brain development!
During pregnancy, it helps prevent neural tube defects and plays a big role in forming a normal and healthy baby’s brain and spinal cord. Folate also helps cells divide and assists in both DNA and RNA synthesis.
Emerging research suggests that the presence of FRAAs negatively impacts folate transport into the brain.
- Recent studies reveal that a large subgroup of children with autism spectrum disorder (ASD) have FRAAs.
- This suggests that a possible disruption in folate transport across the blood-cerebrospinal fluid (CSF) barrier may potentially influence ASD-linked brain development.
- Screening for the FRAAs in your child should be part of your early intervention strategies.
Is there a test for identifying Folate Receptor Autoantibodies (FRAAs)?
Yes, there is a test – The Folate Receptor Antibody Test (FRAT®) has emerged as a diagnostic tool for detecting the presence of FRAAs.
It is important to screen at an early age or as soon as possible as there may be corrective measures available. Please consult your physician for further information.
To request a test kit, click on the button below.

For information on autism monitoring, screening and testing please read our blog.
References
- Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, Gropman AL, Ashwal S. Evidence report: Genetic and metabolic testing on children with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2011 Oct 25;77(17):1629-35. doi: 10.1212/WNL.0b013e3182345896. Epub 2011 Sep 28. PMID: 21956720.
https://pubmed.ncbi.nlm.nih.gov/21956720/ - Lipkin PH, Macias MM; COUNCIL ON CHILDREN WITH DISABILITIES, SECTION ON DEVELOPMENTAL AND BEHAVIORAL PEDIATRICS. Promoting Optimal Development: Identifying Infants and Young Children With Developmental Disorders Through Developmental Surveillance and Screening. Pediatrics. 2020 Jan;145(1):e20193449. doi: 10.1542/peds.2019-3449. Epub 2019 Dec 16. PMID: 31843861.
https://pubmed.ncbi.nlm.nih.gov/31843861/ - Choo YY, Agarwal P, How CH, Yeleswarapu SP. Developmental delay: identification and management at primary care level. Singapore Med J. 2019 Mar;60(3):119-123. doi: 10.11622/smedj.2019025. PMID: 30997518; PMCID: PMC6441684.
https://pubmed.ncbi.nlm.nih.gov/30997518/ - Frye RE. Metabolic and mitochondrial disorders associated with epilepsy in children with autism spectrum disorder. Epilepsy Behav. 2015 Jun;47:147-57. doi: 10.1016/j.yebeh.2014.08.134. Epub 2014 Nov 4. PMID: 25440829.
https://pubmed.ncbi.nlm.nih.gov/25440829/ - Ogier de Baulny H, Saudubray JM. Branched-chain organic acidurias. Semin Neonatol. 2002 Feb;7(1):65-74. doi: 10.1053/siny.2001.0087. PMID: 12069539.
https://pubmed.ncbi.nlm.nih.gov/12069539/ - Dionisi-Vici C, Deodato F, Röschinger W, Rhead W, Wilcken B. ‘Classical’ organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: long-term outcome and effects of expanded newborn screening using tandem mass spectrometry. J Inherit Metab Dis. 2006 Apr-Jun;29(2-3):383-9. doi: 10.1007/s10545-006-0278-z. PMID: 16763906.
https://pubmed.ncbi.nlm.nih.gov/16763906/ - van de Burgt N, van Doesum W, Grevink M, van Niele S, de Koning T, Leibold N, Martinez-Martinez P, van Amelsvoort T, Cath D. Psychiatric manifestations of inborn errors of metabolism: A systematic review. Neurosci Biobehav Rev. 2023 Jan;144:104970. doi: 10.1016/j.neubiorev.2022.104970. Epub 2022 Nov 25. PMID: 36436739.
https://pubmed.ncbi.nlm.nih.gov/36436739/



Comments are closed.